Department of Neurology, Columbia University Medical Center, New York City, New York, USA.
MRC Mitochondrial Biology Unit, Cambridge Biomedical Campus, Cambridge, UK.
J Med Genet. 2018 Aug;55(8):515-521. doi: 10.1136/jmedgenet-2017-105012. Epub 2018 Mar 30.
Thymine kinase 2 (TK2) is a mitochondrial matrix protein encoded in nuclear DNA and phosphorylates the pyrimidine nucleosides: thymidine and deoxycytidine. Autosomal recessive mutations cause a spectrum of disease from infantile onset to adult onset manifesting primarily as myopathy.
To perform a retrospective natural history study of a large cohort of patients with TK2 deficiency.
The study was conducted by 42 investigators across 31 academic medical centres.
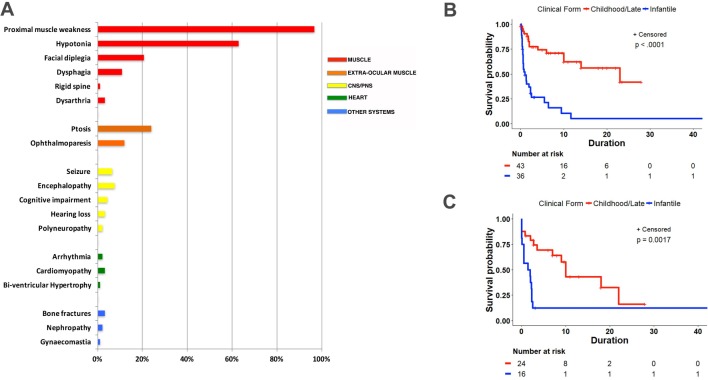
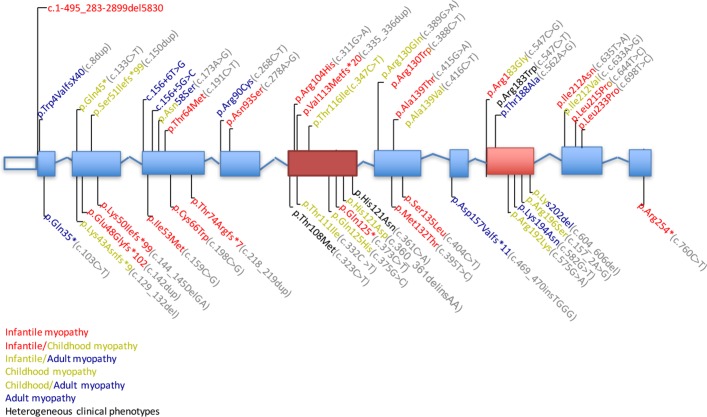
We identified 92 patients with genetically confirmed diagnoses of TK2 deficiency: 67 from literature review and 25 unreported cases. Based on clinical and molecular genetics findings, we recognised three phenotypes with divergent survival: (1) infantile-onset myopathy (42.4%) with severe mitochondrial DNA (mtDNA) depletion, frequent neurological involvement and rapid progression to early mortality (median post-onset survival (POS) 1.00, CI 0.58 to 2.33 years); (2) childhood-onset myopathy (40.2%) with mtDNA depletion, moderate-to-severe progression of generalised weakness and median POS at least 13 years; and (3) late-onset myopathy (17.4%) with mild limb weakness at onset and slow progression to respiratory insufficiency with median POS of 23 years. Ophthalmoparesis and facial weakness are frequent in adults. Muscle biopsies show multiple mtDNA deletions often with mtDNA depletion.
In TK2 deficiency, age at onset, rate of weakness progression and POS are important variables that define three clinical subtypes. Nervous system involvement often complicates the clinical course of the infantile-onset form while extraocular muscle and facial involvement are characteristic of the late-onset form. Our observations provide essential information for planning future clinical trials in this disorder.
胸腺激酶 2(TK2)是一种位于线粒体基质中的核 DNA 编码蛋白,可磷酸化嘧啶核苷:胸苷和脱氧胞苷。常染色体隐性突变导致从婴儿期到成年期发病的一系列疾病,主要表现为肌病。
对大量 TK2 缺乏症患者进行回顾性自然史研究。
该研究由 42 名来自 31 个学术医疗中心的调查员进行。
我们鉴定了 92 名经基因确诊的 TK2 缺乏症患者:67 名来自文献回顾,25 名未报告病例。基于临床和分子遗传学发现,我们识别出三种具有不同生存趋势的表型:(1)婴儿期起病的肌病(42.4%),表现为严重的线粒体 DNA(mtDNA)耗竭、频繁的神经系统受累和快速进展至早期死亡(中位发病后生存(POS)1.00,CI 0.58 至 2.33 年);(2)儿童期起病的肌病(40.2%),mtDNA 耗竭,全身性肌无力呈中重度进展,中位 POS 至少 13 年;(3)迟发性肌病(17.4%),起病时仅表现为轻度肢体无力,进展缓慢导致呼吸功能不全,中位 POS 为 23 年。眼肌麻痹和面肌无力在成人中常见。肌肉活检显示多种 mtDNA 缺失,常伴有 mtDNA 耗竭。
在 TK2 缺乏症中,发病年龄、肌无力进展速度和 POS 是定义三种临床亚型的重要变量。神经系统受累常使婴儿期起病形式的临床病程复杂化,而眼外肌和面部受累是迟发性起病形式的特征。我们的观察结果为该疾病的未来临床试验提供了重要信息。