Center for Microbial Ecology and Technology (CMET), Ghent University, Coupure Links 653, B-9000, Ghent, Belgium.
Infrastructure and Environment Research Division, School of Engineering, University of Glasgow, Rankine Building, Oakfield Avenue, Glasgow, G12 8LT, UK.
Microbiome. 2018 Apr 2;6(1):63. doi: 10.1186/s40168-018-0449-9.
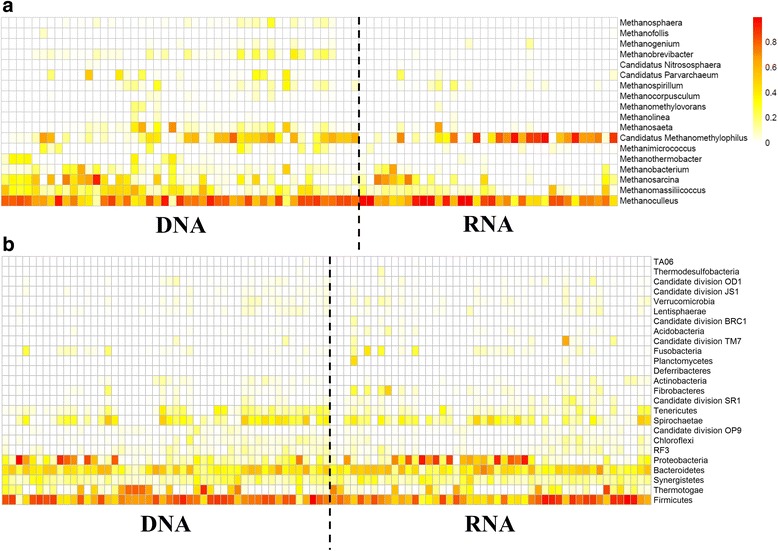
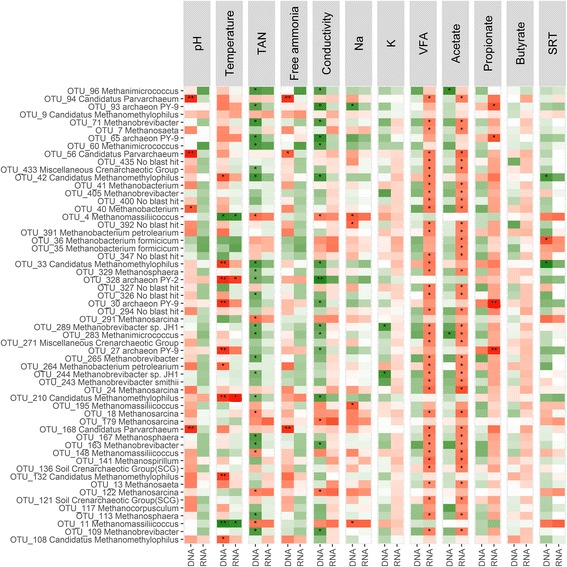
Amplicon sequencing methods targeting the 16S rRNA gene have been used extensively to investigate microbial community composition and dynamics in anaerobic digestion. These methods successfully characterize amplicons but do not distinguish micro-organisms that are actually responsible for the process. In this research, the archaeal and bacterial community of 48 full-scale anaerobic digestion plants were evaluated on DNA (total community) and RNA (active community) level via 16S rRNA (gene) amplicon sequencing.
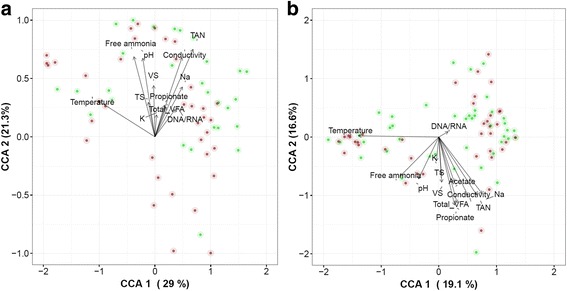
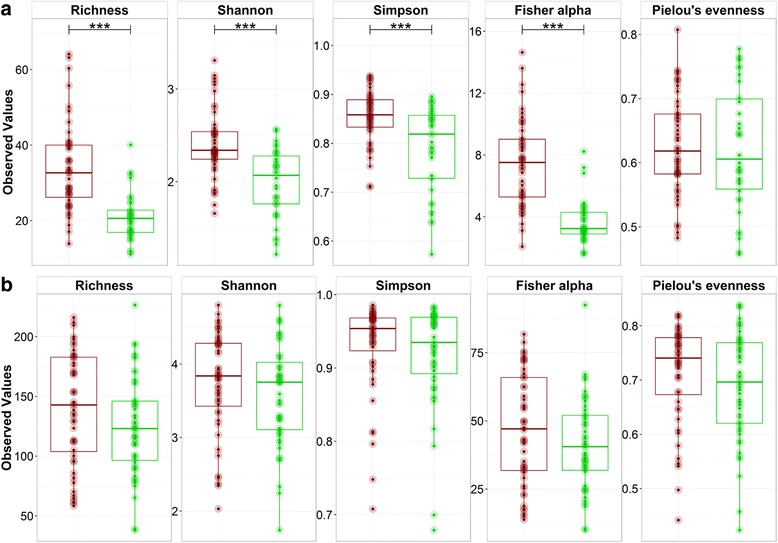
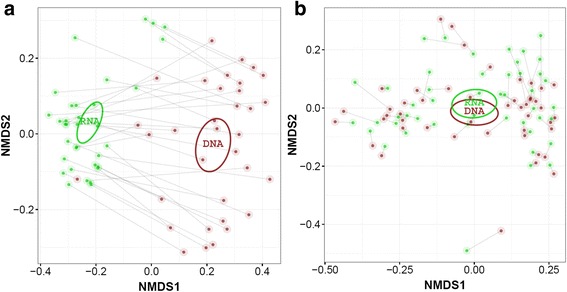
A significantly higher diversity on DNA compared with the RNA level was observed for archaea, but not for bacteria. Beta diversity analysis showed a significant difference in community composition between the DNA and RNA of both bacteria and archaea. This related with 25.5 and 42.3% of total OTUs for bacteria and archaea, respectively, that showed a significant difference in their DNA and RNA profiles. Similar operational parameters affected the bacterial and archaeal community, yet the differentiating effect between DNA and RNA was much stronger for archaea. Co-occurrence networks and functional prediction profiling confirmed the clear differentiation between DNA and RNA profiles.
In conclusion, a clear difference in active (RNA) and total (DNA) community profiles was observed, implying the need for a combined approach to estimate community stability in anaerobic digestion.
针对 16S rRNA 基因的扩增子测序方法已被广泛用于研究厌氧消化中的微生物群落组成和动态变化。这些方法可以成功地对扩增子进行特征描述,但无法区分实际参与过程的微生物。在这项研究中,通过 16S rRNA(基因)扩增子测序,对 48 个全规模厌氧消化厂的古菌和细菌群落的 DNA(总群落)和 RNA(活性群落)水平进行了评估。
与 RNA 水平相比,古菌的 DNA 多样性明显更高,但细菌则不然。β多样性分析显示,细菌和古菌的 DNA 和 RNA 之间的群落组成存在显著差异。这与细菌和古菌的总 OTUs 的 25.5%和 42.3%分别相关,它们的 DNA 和 RNA 图谱存在显著差异。类似的操作参数影响了细菌和古菌群落,但古菌的 DNA 和 RNA 之间的区分效果要强得多。共现网络和功能预测分析证实了 DNA 和 RNA 图谱之间的明显差异。
总之,观察到了活跃(RNA)和总(DNA)群落图谱之间的明显差异,这意味着需要采用综合方法来估计厌氧消化中的群落稳定性。