Department of Applied Physics, Columbia University, New York, NY 10027.
Department of Systems Biology, Columbia University, New York, NY 10032.
Proc Natl Acad Sci U S A. 2018 Apr 17;115(16):E3692-E3701. doi: 10.1073/pnas.1714376115. Epub 2018 Apr 2.
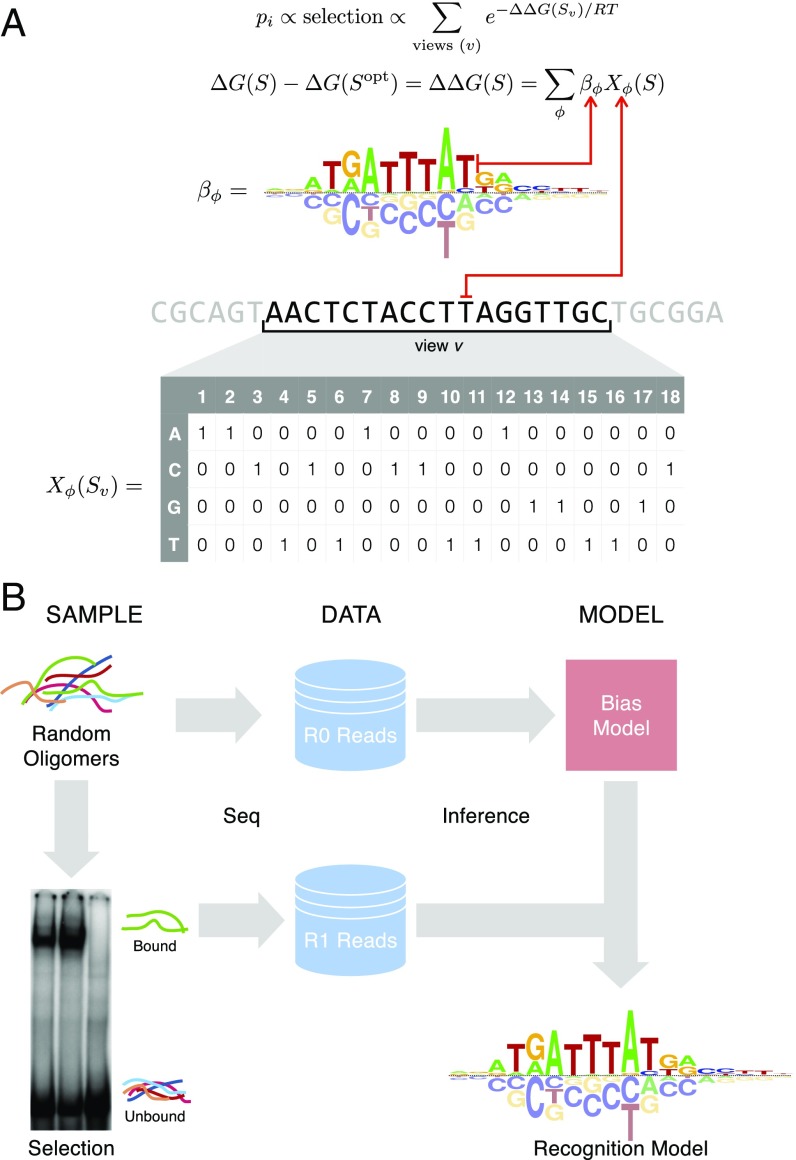
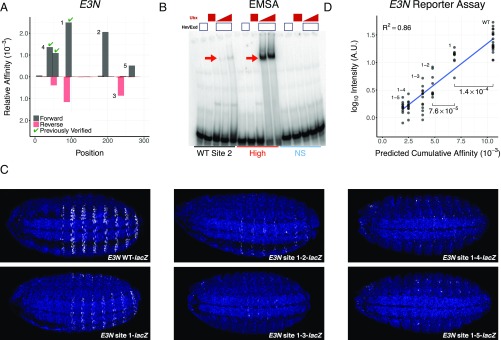
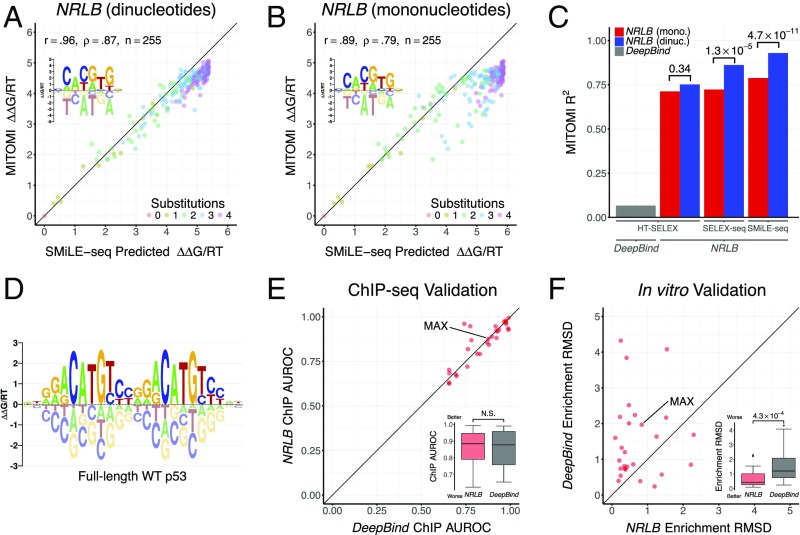
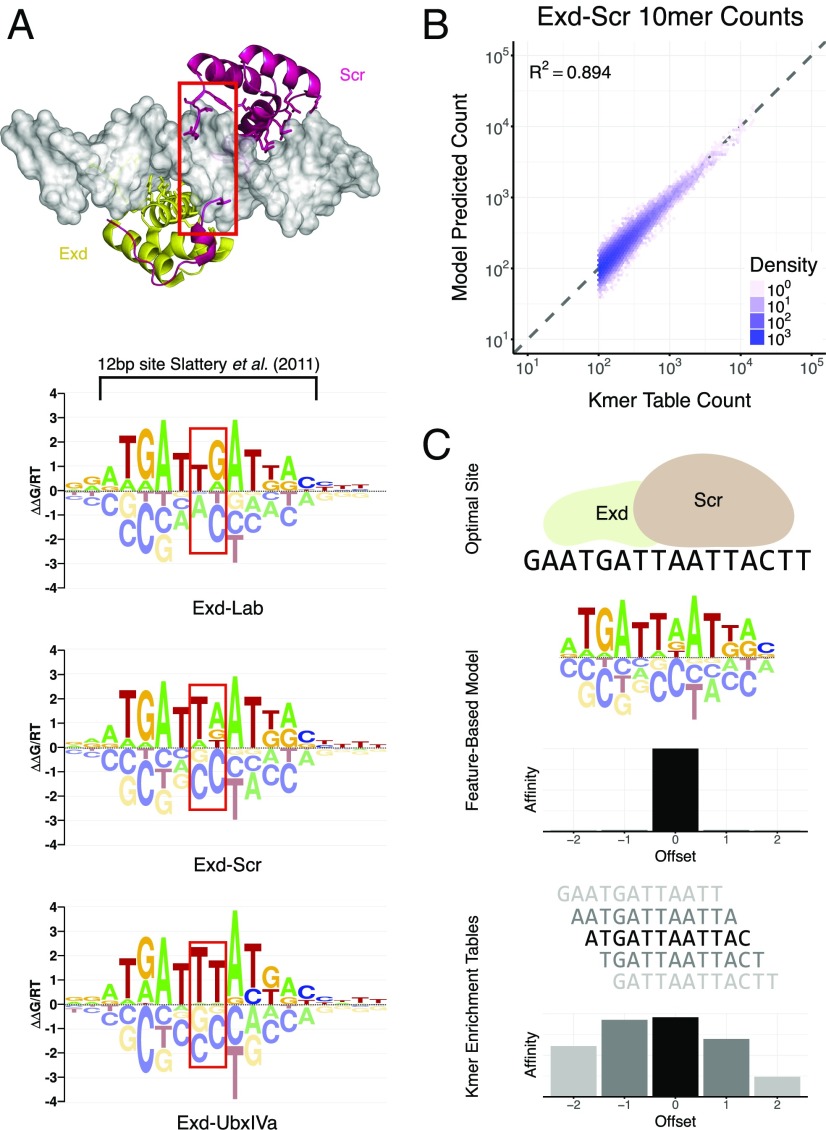
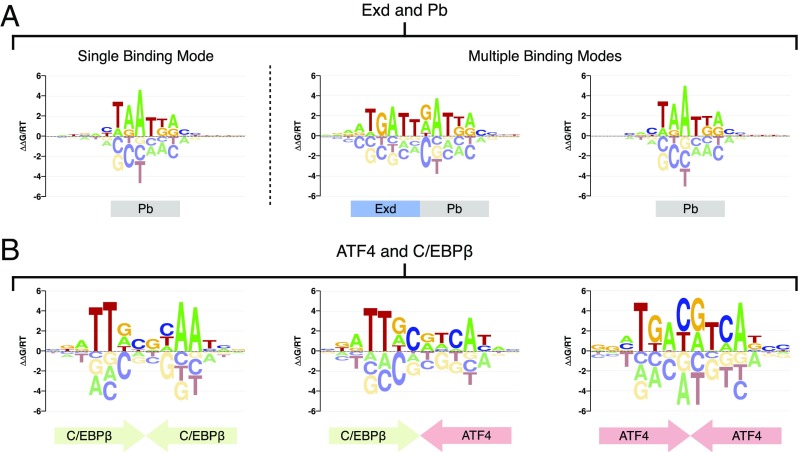
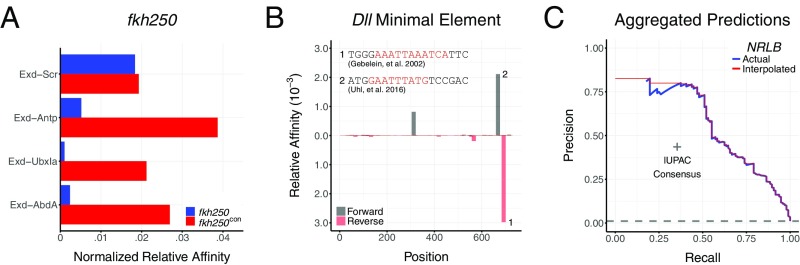
Transcription factors (TFs) control gene expression by binding to genomic DNA in a sequence-specific manner. Mutations in TF binding sites are increasingly found to be associated with human disease, yet we currently lack robust methods to predict these sites. Here, we developed a versatile maximum likelihood framework named No Read Left Behind (NRLB) that infers a biophysical model of protein-DNA recognition across the full affinity range from a library of in vitro selected DNA binding sites. NRLB predicts human Max homodimer binding in near-perfect agreement with existing low-throughput measurements. It can capture the specificity of the p53 tetramer and distinguish multiple binding modes within a single sample. Additionally, we confirm that newly identified low-affinity enhancer binding sites are functional in vivo, and that their contribution to gene expression matches their predicted affinity. Our results establish a powerful paradigm for identifying protein binding sites and interpreting gene regulatory sequences in eukaryotic genomes.
转录因子 (TFs) 通过与基因组 DNA 以序列特异性的方式结合来控制基因表达。越来越多的研究发现,TF 结合位点的突变与人类疾病有关,但我们目前缺乏可靠的方法来预测这些位点。在这里,我们开发了一种通用的最大似然框架,名为 No Read Left Behind (NRLB),该框架可以从体外选择的 DNA 结合位点库中推断出蛋白质-DNA 识别的生物物理模型,涵盖整个亲和力范围。NRLB 预测人类 Max 同源二聚体的结合与现有低通量测量结果几乎完全一致。它可以捕捉 p53 四聚体的特异性,并在单个样本中区分多种结合模式。此外,我们证实新鉴定的低亲和力增强子结合位点在体内具有功能,并且它们对基因表达的贡献与其预测的亲和力相匹配。我们的研究结果为识别蛋白质结合位点和解释真核基因组中的基因调控序列建立了一个强大的范例。