Yan Muqing, Byrne David H, Klein Patricia E, Yang Jizhou, Dong Qianni, Anderson Natalie
1Department of Horticultural Sciences, Texas A&M University, College Station, TX 77843 USA.
2Institute for Plant Genomics and Biotechnology, Texas A&M University, College Station, TX 77843 USA.
Hortic Res. 2018 Apr 1;5:17. doi: 10.1038/s41438-018-0021-6. eCollection 2018.
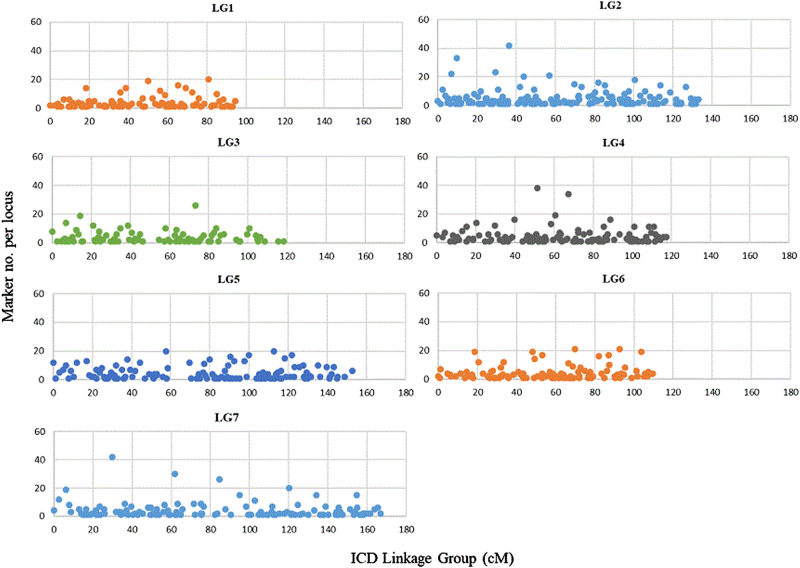
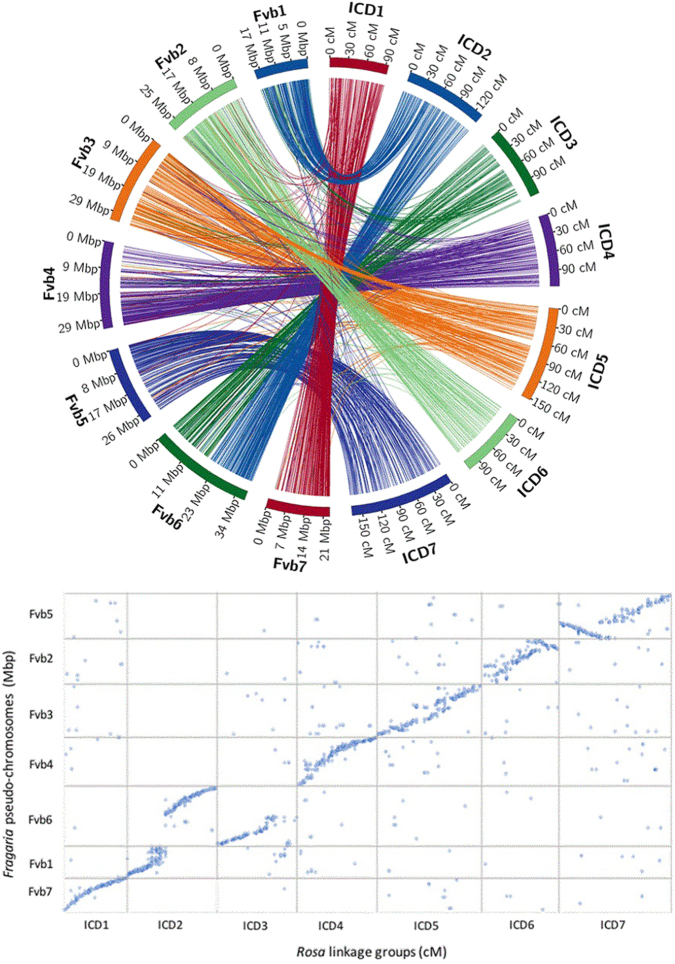
Roses, which have been cultivated for at least 5000 years, are one of the most important ornamental crops in the world. Because of the interspecific nature and high heterozygosity in commercial roses, the genetic resources available for rose are limited. To effectively identify markers associated with QTL controlling important traits, such as disease resistance, abundant markers along the genome and careful phenotyping are required. Utilizing genotyping by sequencing technology and the strawberry genome ( v2.0.a1) as a reference, we generated thousands of informative single nucleotide polymorphism (SNP) markers. These SNPs along with known bridge simple sequence repeat (SSR) markers allowed us to create the first high-density integrated consensus map for diploid roses. Individual maps were first created for populations J06-20-14-3×"Little Chief" (J14-3×LC), J06-20-14-3×"Vineyard Song" (J14-3×VS) and "Old Blush"×"Red Fairy" (OB×RF) and these maps were linked with 824 SNPs and 13 SSR bridge markers. The anchor SSR markers were used to determine the numbering of the rose linkage groups. The diploid consensus map has seven linkage groups (LGs), a total length of 892.2 cM, and an average distance of 0.25 cM between 3527 markers. By combining three individual populations, the marker density and the reliability of the marker order in the consensus map was improved over a single population map. Extensive synteny between the strawberry and diploid rose genomes was observed. This consensus map will serve as the tool for the discovery of marker-trait associations in rose breeding using pedigree-based analysis. The high level of conservation observed between the strawberry and rose genomes will help further comparative studies within the Rosaceae family and may aid in the identification of candidate genes within QTL regions.
玫瑰的种植历史至少已有5000年,是世界上最重要的观赏作物之一。由于商业玫瑰的种间特性和高杂合性,可用于玫瑰的遗传资源有限。为了有效鉴定与控制重要性状(如抗病性)的数量性状基因座(QTL)相关的标记,需要全基因组丰富的标记以及细致的表型分析。利用测序技术进行基因分型,并以草莓基因组(v2.0.a1)作为参考,我们生成了数千个信息丰富的单核苷酸多态性(SNP)标记。这些SNP与已知的桥梁简单序列重复(SSR)标记一起,使我们能够创建第一张二倍体玫瑰的高密度整合共识图谱。首先为群体J06 - 20 - 14 - 3דLittle Chief”(J14 - 3×LC)、J06 - 20 - 14 - 3דVineyard Song”(J14 - 3×VS)和“Old Blush”דRed Fairy”(OB×RF)创建了个体图谱,这些图谱通过824个SNP和13个SSR桥梁标记进行了连锁。锚定SSR标记用于确定玫瑰连锁群的编号。二倍体共识图谱有7个连锁群(LGs),总长度为892.2 cM,3527个标记之间的平均距离为0.25 cM。通过合并三个个体群体,共识图谱中的标记密度和标记顺序的可靠性比单个群体图谱有所提高。观察到草莓和二倍体玫瑰基因组之间存在广泛的共线性。这一共识图谱将作为利用基于系谱分析的工具,用于发现玫瑰育种中标记 - 性状关联。草莓和玫瑰基因组之间观察到的高度保守性将有助于蔷薇科内进一步的比较研究,并可能有助于鉴定QTL区域内的候选基因。