Department of Bioengineering, California Institute of Technology, Pasadena, United States.
Department of Applied Physics, California Institute of Technology, Pasadena, United States.
Elife. 2018 Apr 19;7:e31955. doi: 10.7554/eLife.31955.
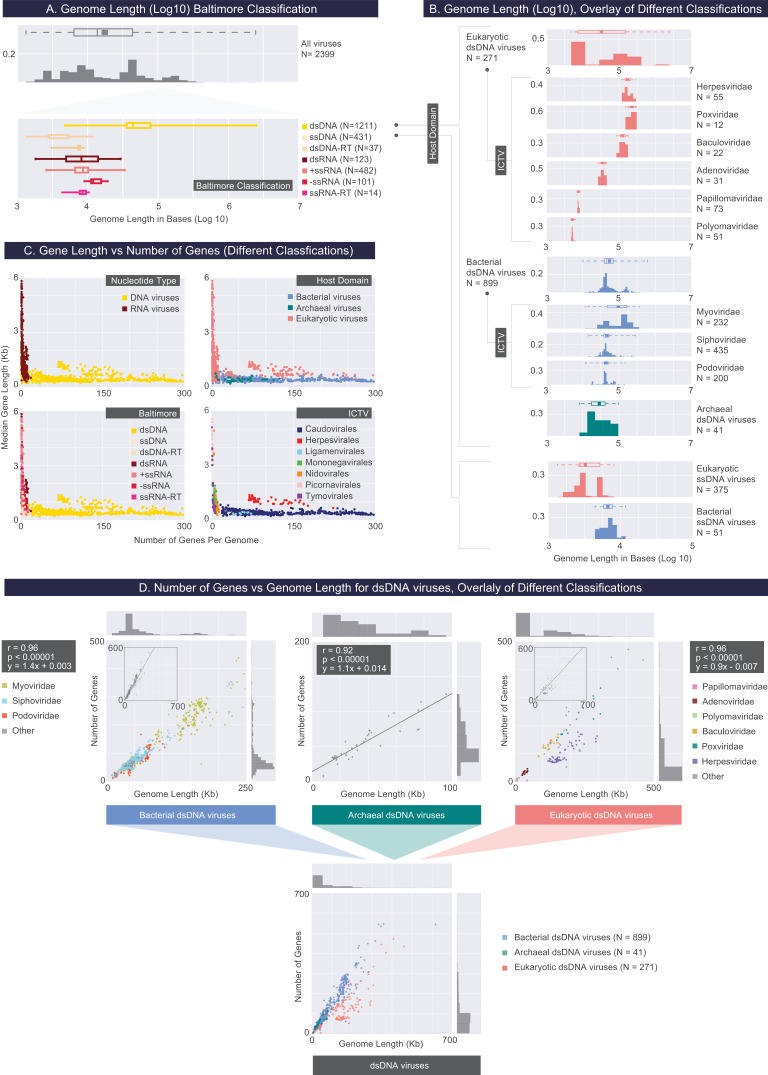
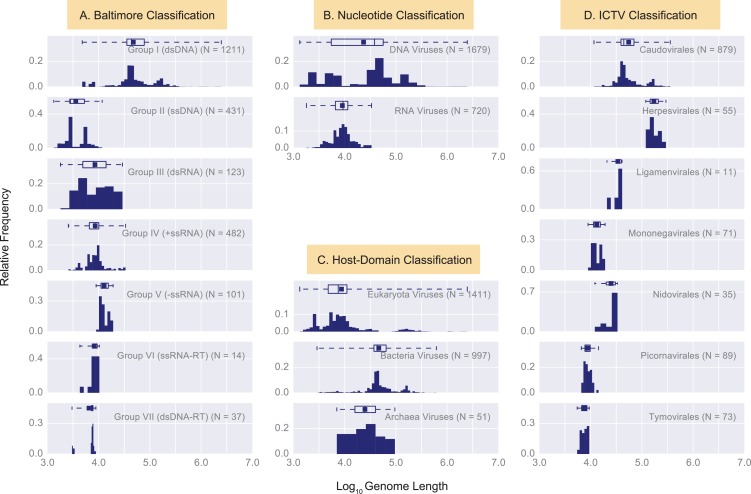
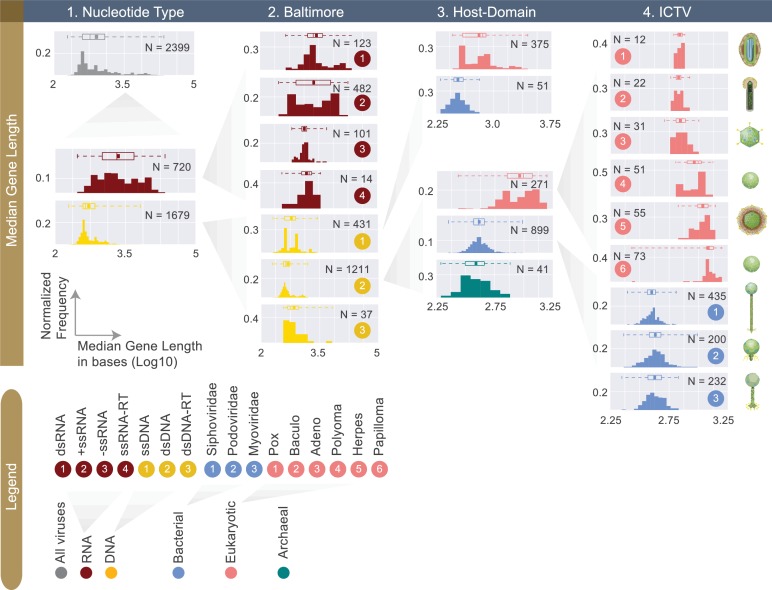
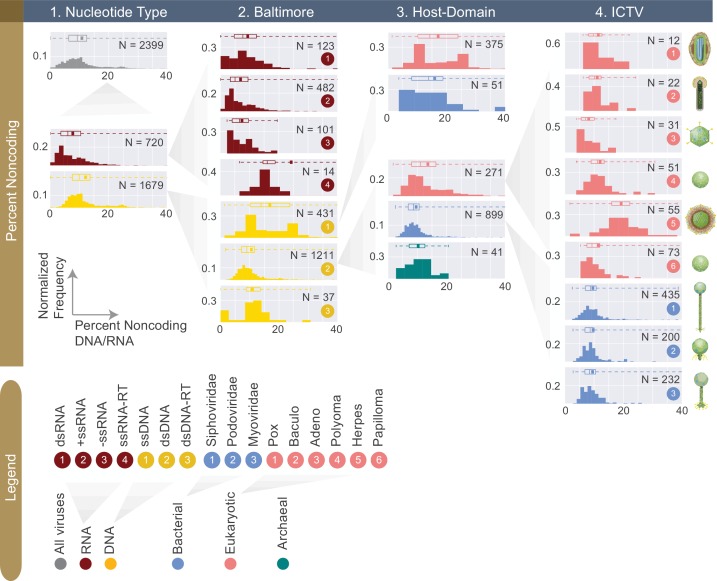
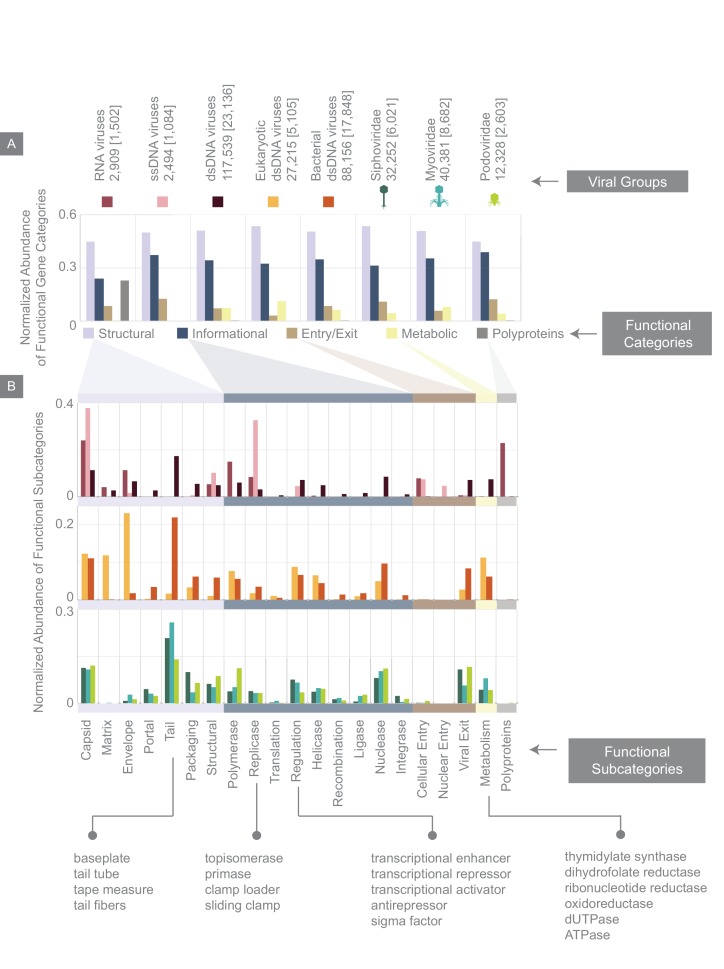
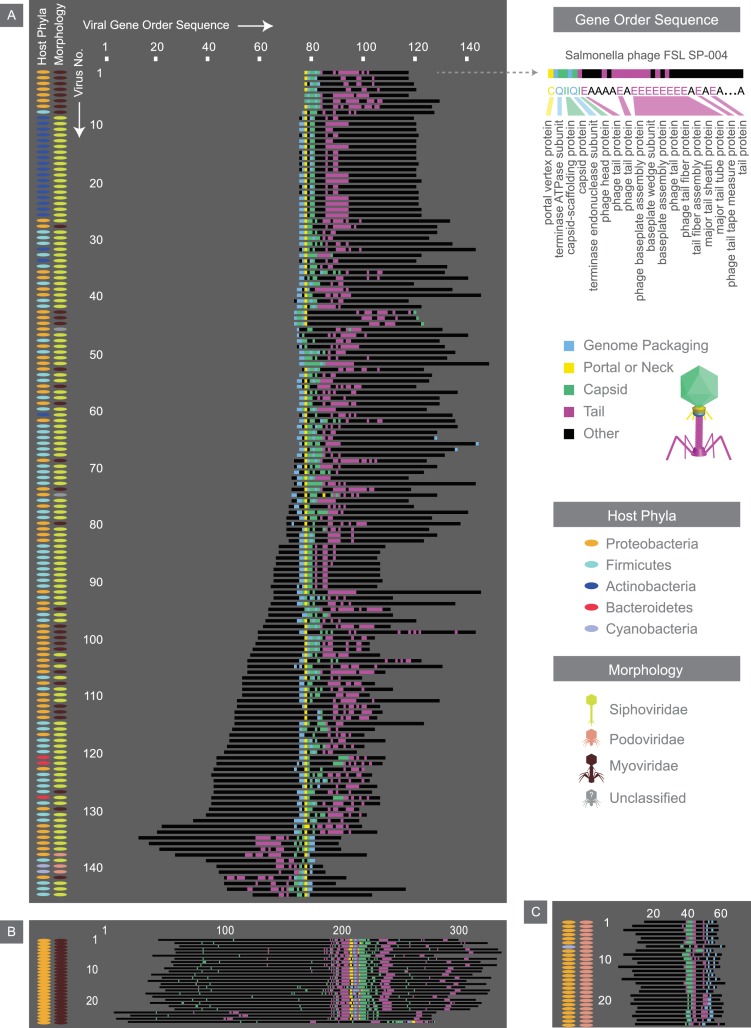
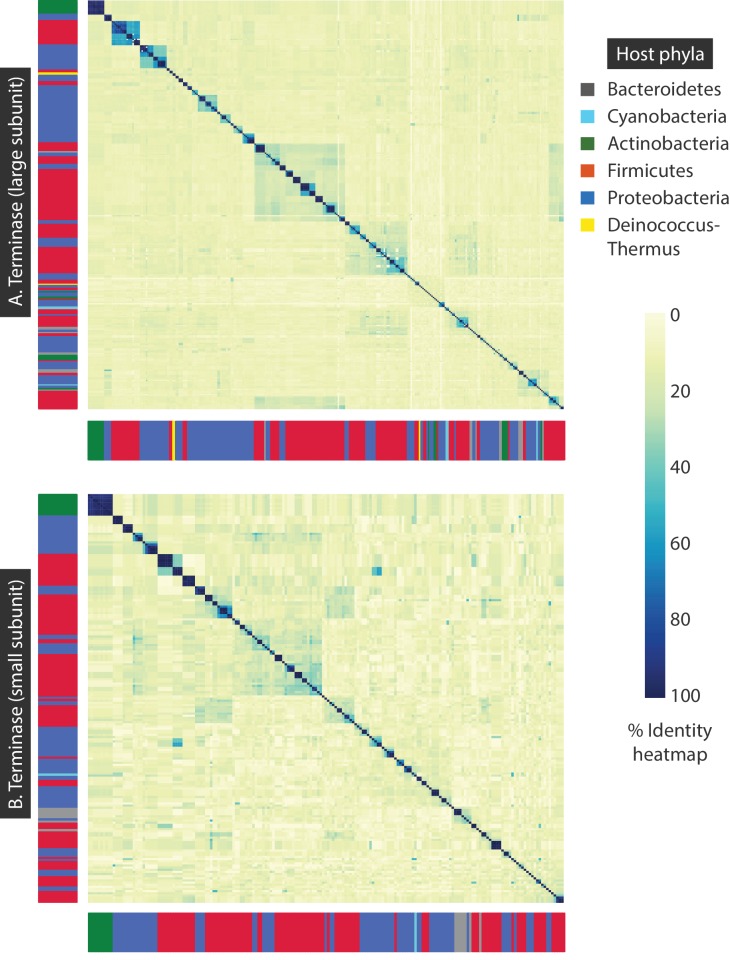
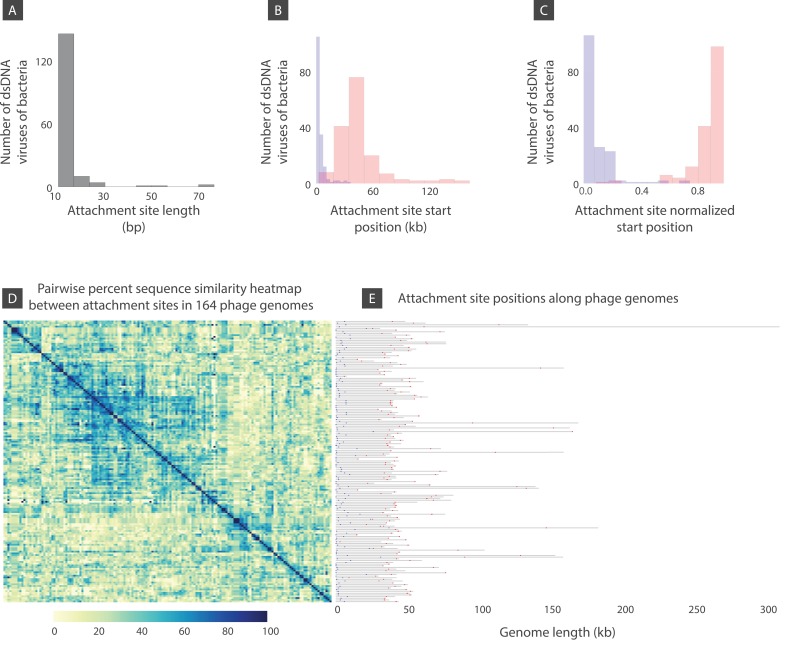
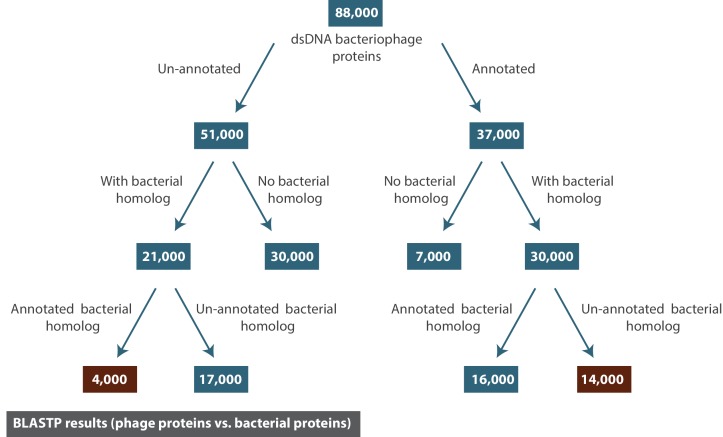
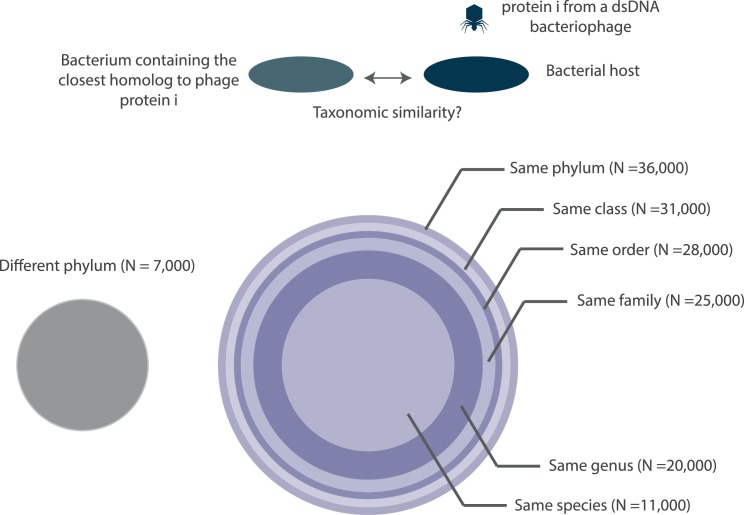
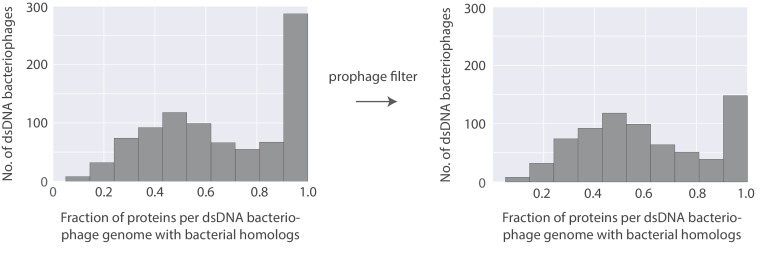
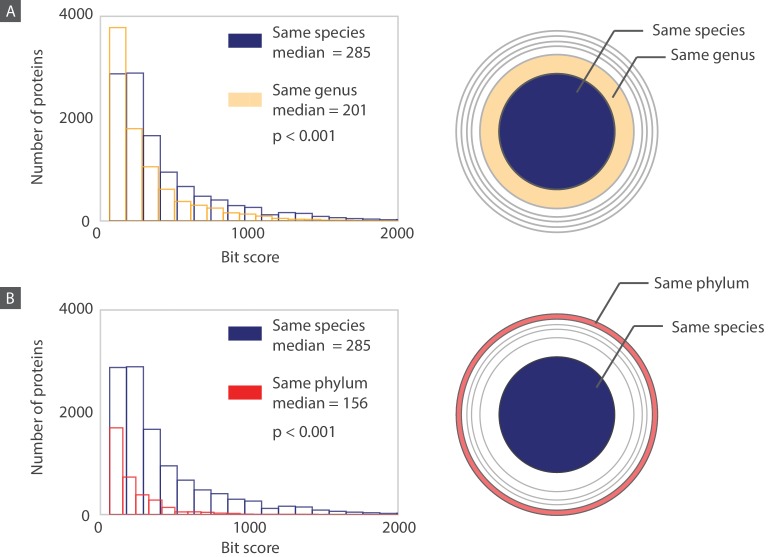
The complete assembly of viral genomes from metagenomic datasets (short genomic sequences gathered from environmental samples) has proven to be challenging, so there are significant blind spots when we view viral genomes through the lens of metagenomics. One approach to overcoming this problem is to leverage the thousands of complete viral genomes that are publicly available. Here we describe our efforts to assemble a comprehensive resource that provides a quantitative snapshot of viral genomic trends - such as gene density, noncoding percentage, and abundances of functional gene categories - across thousands of viral genomes. We have also developed a coarse-grained method for visualizing viral genome organization for hundreds of genomes at once, and have explored the extent of the overlap between bacterial and bacteriophage gene pools. Existing viral classification systems were developed prior to the sequencing era, so we present our analysis in a way that allows us to assess the utility of the different classification systems for capturing genomic trends.
从宏基因组数据集(从环境样本中收集的短基因组序列)中完整组装病毒基因组一直具有挑战性,因此,通过宏基因组学的视角观察病毒基因组时存在很大的盲点。克服这个问题的一种方法是利用成千上万种公开可用的完整病毒基因组。在这里,我们描述了我们努力组装一个全面的资源,该资源提供了数千种病毒基因组中病毒基因组趋势的定量快照,例如基因密度、非编码百分比和功能基因类别丰度。我们还开发了一种粗粒度的方法,可以一次可视化数百个基因组的病毒基因组组织,并探讨了细菌和噬菌体基因库之间重叠的程度。现有的病毒分类系统是在测序时代之前开发的,因此我们以一种允许我们评估不同分类系统在捕捉基因组趋势方面的效用的方式呈现我们的分析结果。