Daniel Swarovski Research Laboratory, Department of Visceral-, Transplant- and Thoracic Surgery, Medical University of Innsbruck (MUI), Austria.
Mol Oncol. 2018 Jun;12(6):869-882. doi: 10.1002/1878-0261.12199. Epub 2018 May 5.
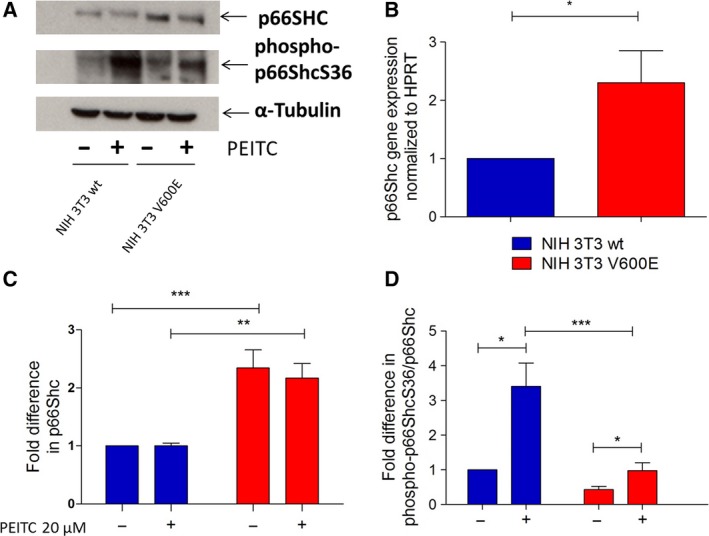
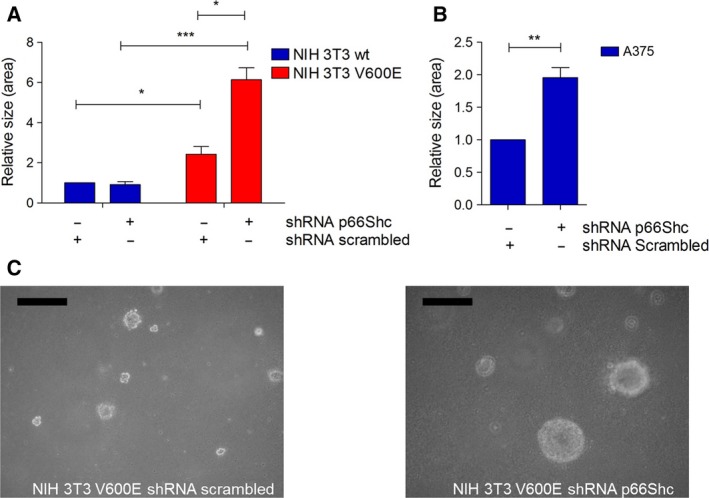
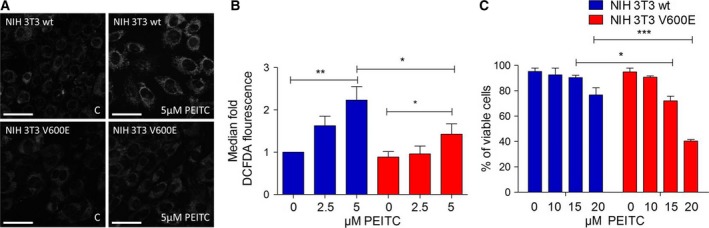
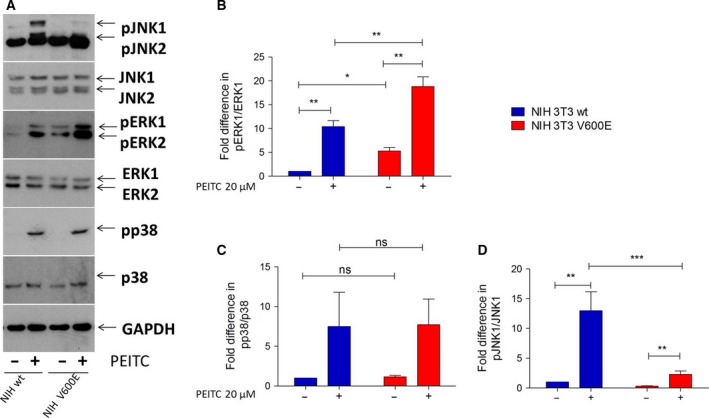
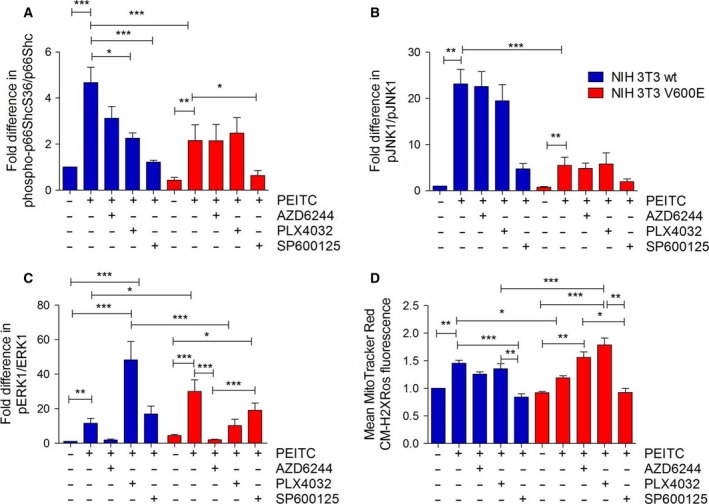
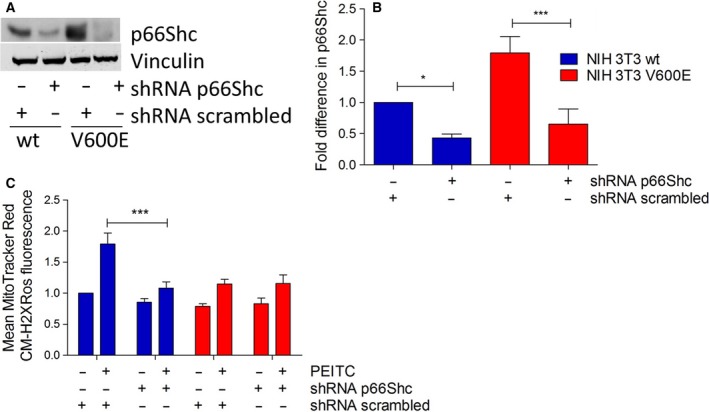
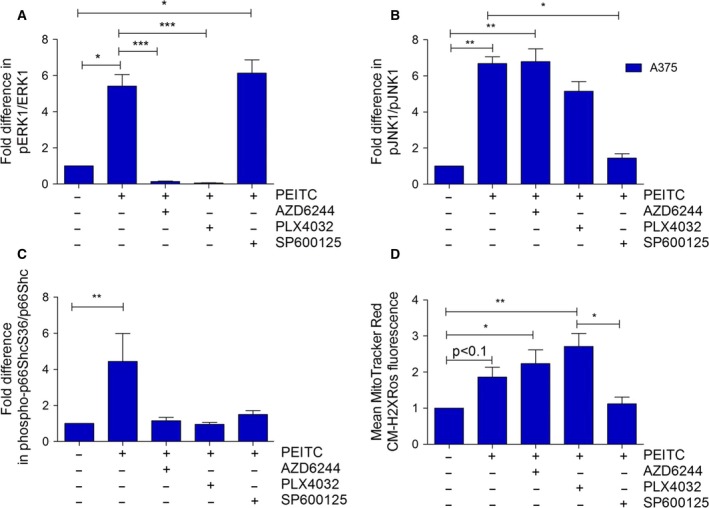
Metabolic reprogramming, as exemplified by the shift from oxidative phosphorylation to glycolysis, is a common feature of transformed cells. In many tumors, altered metabolism is also reflected in increased reactive oxygen species (ROS) levels, which contribute to proliferation and survival signaling. However, despite high ROS levels, cancer cells can be efficiently killed by further increasing ROS production. We have shown previously that both wild-type and oncogenic CRAF and BRAF prevent excessive mitochondrial ROS production. Subsequently, it has been demonstrated that raising ROS levels in BRAFV600E-transformed melanoma cells by inhibiting BRAF or MEK rendered them susceptible to cell death induction. To understand how oncogenic BRAF affects mitochondrial ROS production in melanoma, we studied the mitochondrial ROS-producing oxidoreductase p66Shc, which is frequently overexpressed in tumors. Using NIH 3T3 BRAFV600E fibroblasts and the melanoma cell lines A375 and M238 carrying the same BRAF mutation, we show that under treatment with the ROS-inducing agent phenethyl isothiocyanate (PEITC), oncogenic BRAF renders cells refractory to p66ShcS36 phosphorylation, which is essential for p66Shc activation and mitochondrial ROS production. Consistent with this, the activation of JNK1/2, which phosphorylate S36, was blunted, while other mitogen-activated protein kinases were not affected. Inhibition of JNK1/2 efficiently prevented ROS production, while BRAF and MEK inhibitors increased ROS levels. Vemurafenib-resistant M238R melanoma cells were impaired in S36 phosphorylation and ROS production following PEITC treatment. Moreover, they failed to increase ROS levels after MEK/BRAF inhibition. Finally, shRNA-mediated knockdown of p66Shc led to increased growth of BRAFV600E-transformed NIH 3T3 cells in soft agar assay. Taken together, these data suggest that phosphorylation-activated p66Shc functions as a tumor suppressor in melanoma cells.
代谢重编程,例如从氧化磷酸化到糖酵解的转变,是转化细胞的共同特征。在许多肿瘤中,代谢的改变也反映在活性氧(ROS)水平的升高上,这有助于增殖和存活信号。然而,尽管 ROS 水平很高,但通过进一步增加 ROS 的产生,癌细胞可以被有效地杀死。我们之前已经表明,野生型和致癌 CRAF 和 BRAF 都可以防止线粒体 ROS 的过度产生。随后,研究表明,通过抑制 BRAF 或 MEK 来提高 BRAFV600E 转化的黑素瘤细胞中的 ROS 水平,使它们容易受到细胞死亡诱导。为了了解致癌 BRAF 如何影响黑素瘤中的线粒体 ROS 产生,我们研究了经常在肿瘤中过度表达的线粒体 ROS 产生氧化还原酶 p66Shc。使用 NIH 3T3 BRAFV600E 成纤维细胞和携带相同 BRAF 突变的黑素瘤细胞系 A375 和 M238,我们表明,在 ROS 诱导剂苯乙基异硫氰酸酯(PEITC)的处理下,致癌 BRAF 使细胞对 p66ShcS36 磷酸化产生抗性,这对于 p66Shc 的激活和线粒体 ROS 的产生是必不可少的。与此一致的是,磷酸化 S36 的 JNK1/2 的激活被削弱,而其他丝裂原激活的蛋白激酶不受影响。JNK1/2 的抑制有效地阻止了 ROS 的产生,而 BRAF 和 MEK 抑制剂增加了 ROS 水平。在 PEITC 处理后,vemurafenib 抗性 M238R 黑素瘤细胞中 S36 磷酸化和 ROS 产生受损。此外,它们在 MEK/BRAF 抑制后未能增加 ROS 水平。最后,p66Shc 的 shRNA 介导的敲低导致 BRAFV600E 转化的 NIH 3T3 细胞在软琼脂测定中的生长增加。总之,这些数据表明,磷酸化激活的 p66Shc 在黑素瘤细胞中作为肿瘤抑制因子发挥作用。