Khalid Sana, Drasche Astrid, Thurner Marco, Hermann Martin, Ashraf Muhammad Imtiaz, Fresser Friedrich, Baier Gottfried, Kremser Leopold, Lindner Herbert, Troppmair Jakob
Daniel Swarovski Research Laboratory, Department of Visceral, Transplant and Thoracic Surgery, Medical University of Innsbruck, Innsbruck, Austria.
Department of Anesthesiology and Critical Care Medicine, Medical University of Innsbruck, Innsbruck, Austria.
Sci Rep. 2016 Feb 12;6:20930. doi: 10.1038/srep20930.
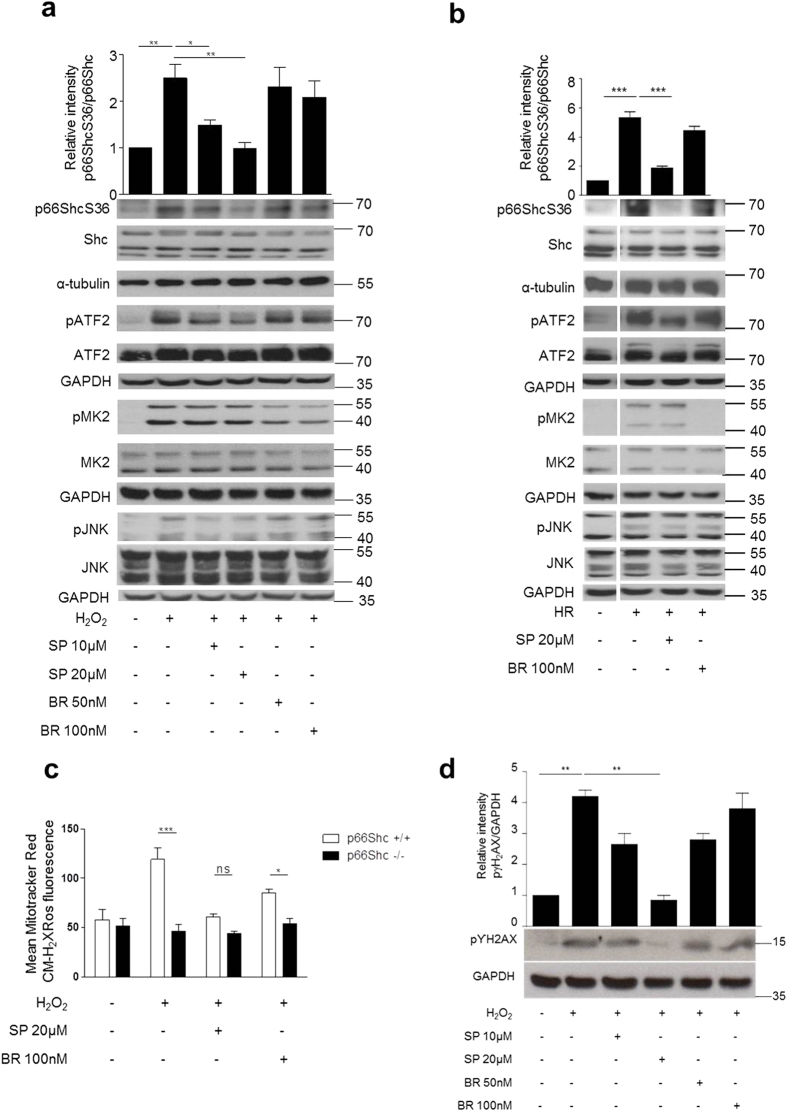
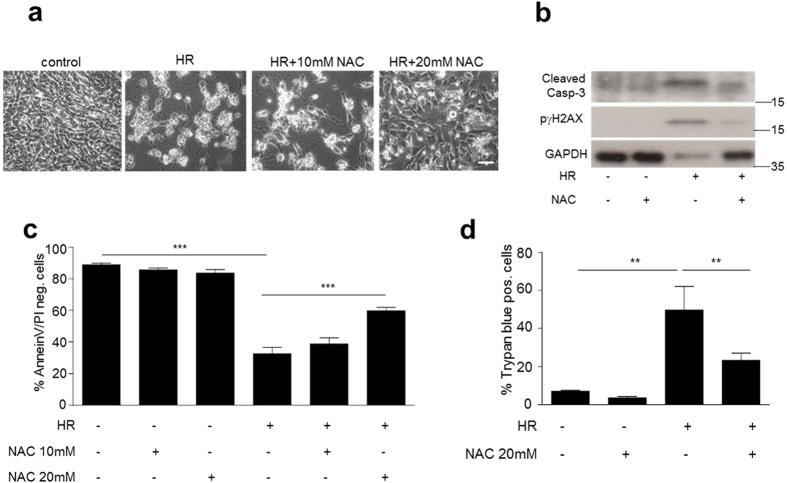
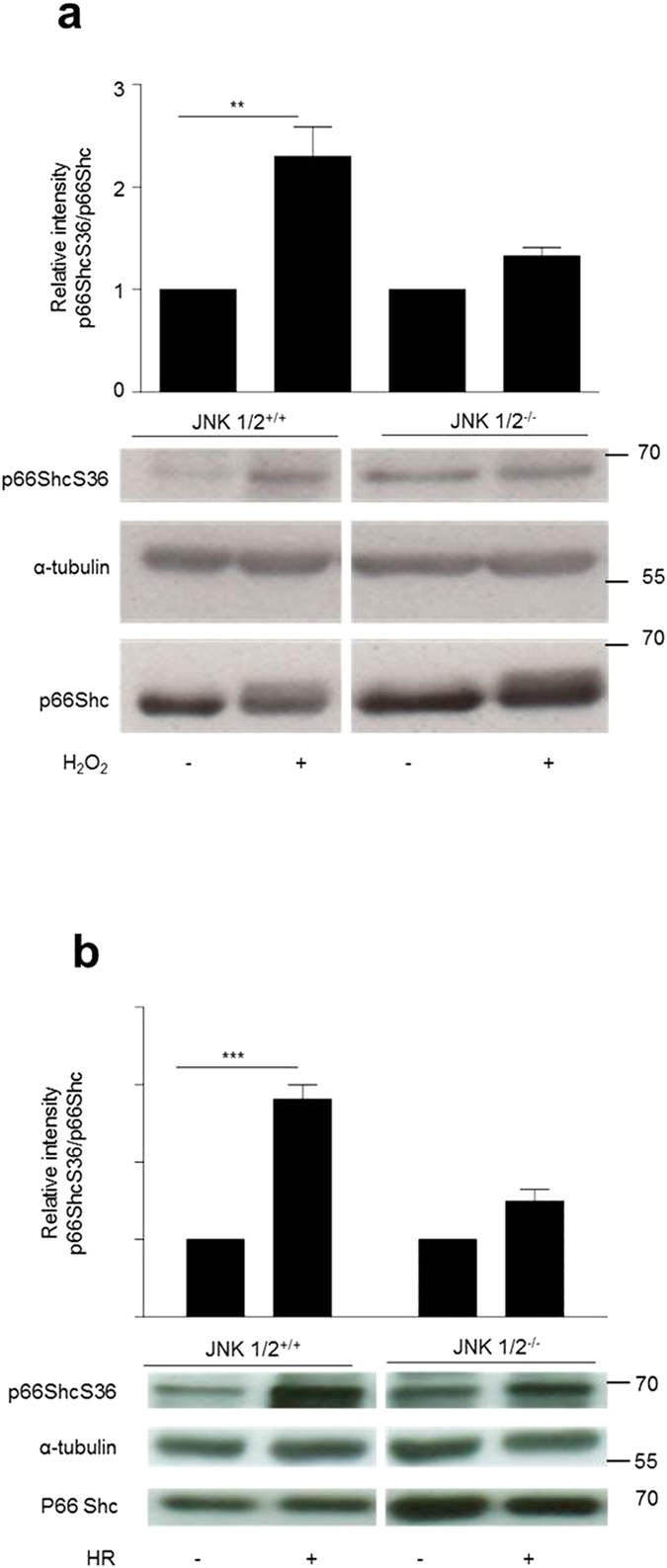
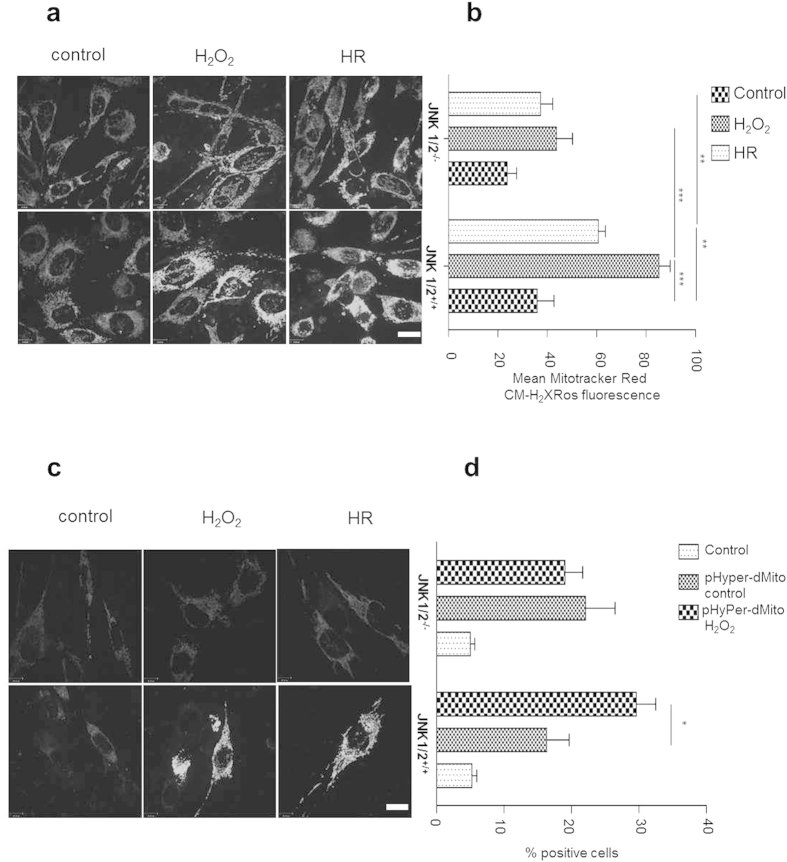
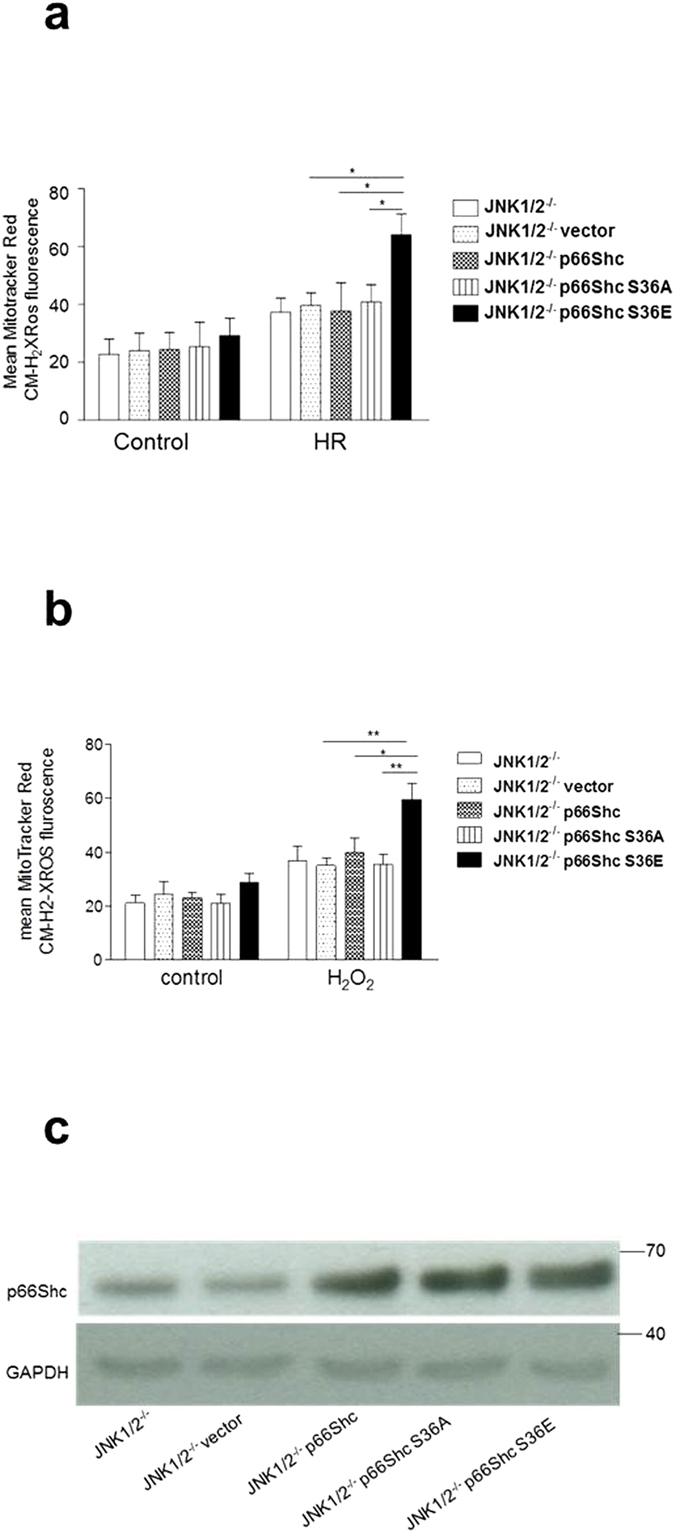
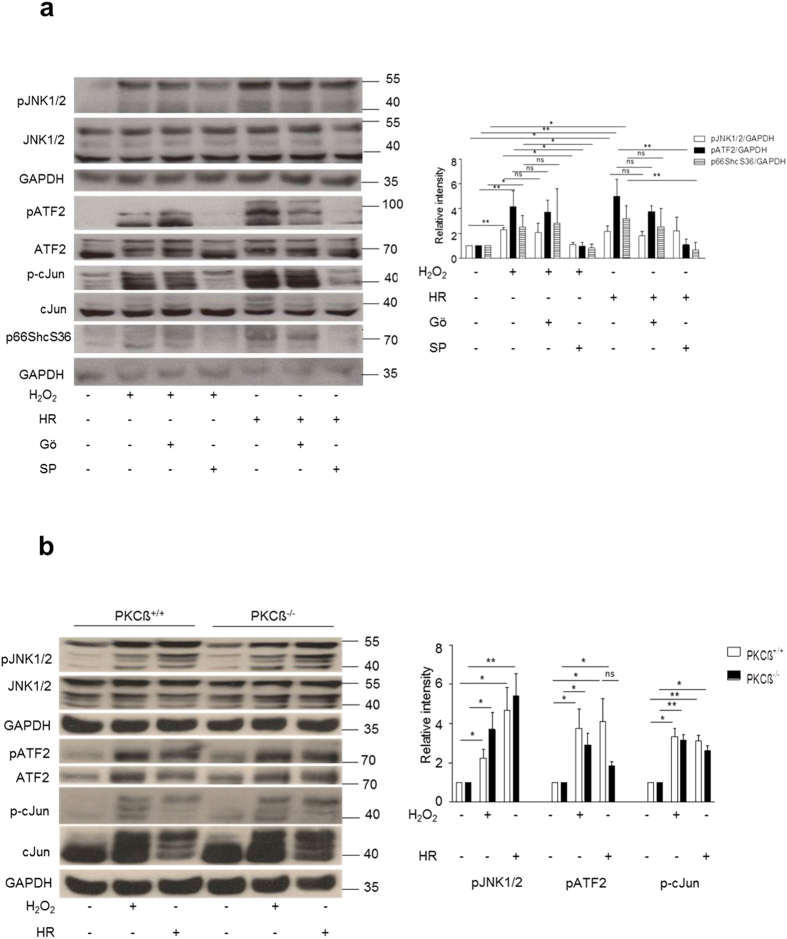
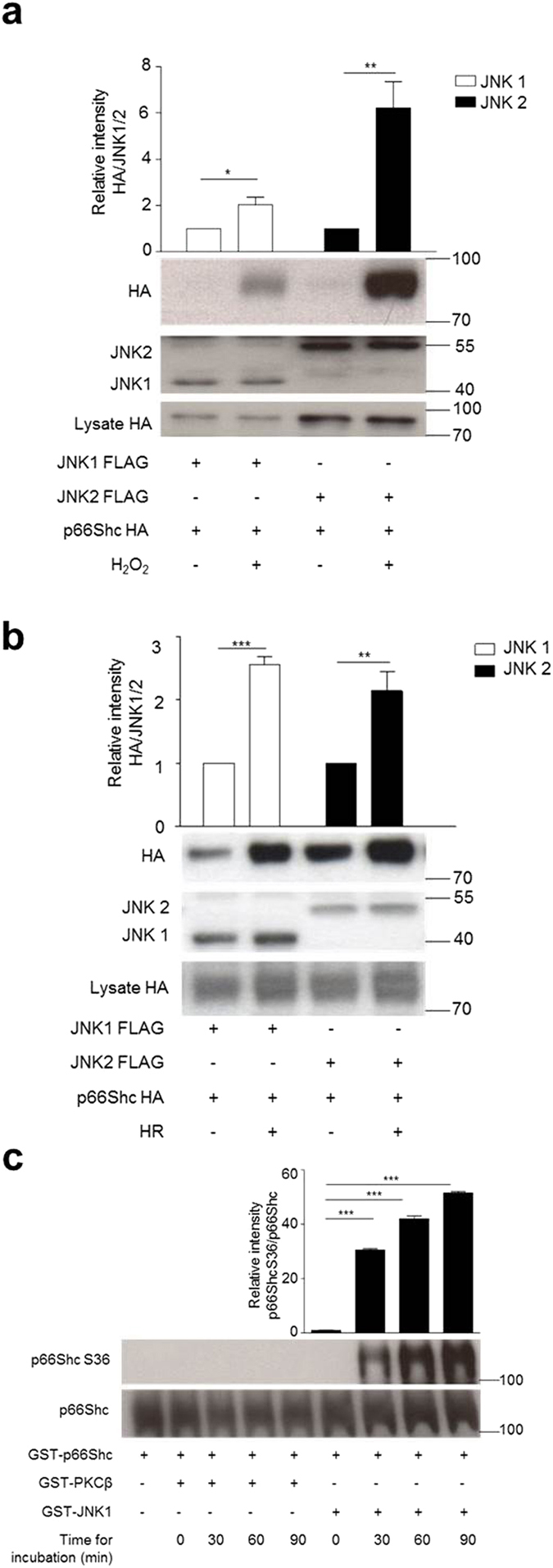
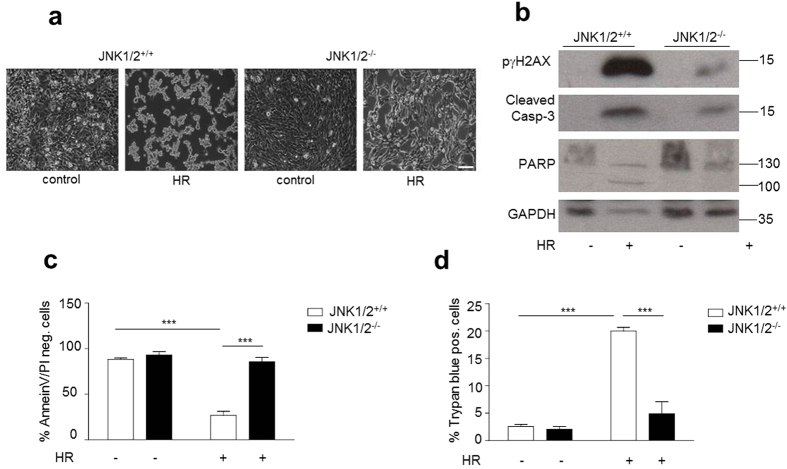
p66Shc-dependent ROS production contributes to many pathologies including ischemia/reperfusion injury (IRI) during solid organ transplantation. Inhibiting p66Shc activation may provide a novel therapeutic approach to prevent damage, which is poorly managed by antioxidants in vivo. Previous work suggested that pro-oxidant and a pro-apoptotic function of p66Shc required mitochondrial import, which depended on serine 36 phosphorylation. PKCß has been proposed as S36 kinase but cJun N-terminal kinases (JNKs) may also phosphorylate this residue. To simulate the early stages of ischemia/reperfusion (IR) we either used H2O2 treatment or hypoxia/reoxygenation (HR). As during reperfusion in vivo, we observed increased JNK and p38 activity in mouse embryonic fibroblasts (MEFs) and HL-1 cardiomyocytes along with significantly increased p66ShcS36 phosphorylation, ROS production and cell damage. Application of specific inhibitors caused a pronounced decrease in p66ShcS36 phosphorylation only in the case of JNK1/2. Moreover, S36 phosphorylation of recombinant p66Shc by JNK1 but not PKCß was demonstrated. We further confirmed JNK1/2-dependent regulation of p66ShcS36 phosphorylation, ROS production and cell death using JNK1/2 deficient MEFs. Finally, the low ROS phenotype of JNK1/2 knockout MEFs was reversed by the phosphomimetic p66ShcS36E mutant. Inhibiting JNK1/2-regulated p66Shc activation may thus provide a therapeutic approach for the prevention of oxidative damage.
p66Shc依赖性活性氧生成参与包括实体器官移植期间的缺血/再灌注损伤(IRI)在内的多种病理过程。抑制p66Shc激活可能提供一种预防损伤的新治疗方法,而抗氧化剂在体内对这种损伤的处理效果不佳。先前的研究表明,p66Shc的促氧化和促凋亡功能需要线粒体导入,这依赖于丝氨酸36磷酸化。蛋白激酶Cβ(PKCβ)被认为是S36激酶,但c-Jun氨基末端激酶(JNKs)也可能使该残基磷酸化。为了模拟缺血/再灌注(IR)的早期阶段,我们要么使用过氧化氢处理,要么使用缺氧/复氧(HR)。正如在体内再灌注期间一样,我们观察到小鼠胚胎成纤维细胞(MEFs)和HL-1心肌细胞中JNK和p38活性增加,同时p66ShcS36磷酸化、活性氧生成和细胞损伤显著增加。应用特异性抑制剂仅在JNK1/2的情况下导致p66ShcS36磷酸化明显降低。此外,证实了JNK1而非PKCβ可使重组p66Shc发生S36磷酸化。我们使用JNK1/2缺陷的MEFs进一步证实了JNK1/2对p66ShcS36磷酸化、活性氧生成和细胞死亡的依赖性调节。最后,磷酸模拟物p66ShcS36E突变体逆转了JNK1/2敲除MEFs的低活性氧表型。因此,抑制JNK1/2调节的p66Shc激活可能为预防氧化损伤提供一种治疗方法。