Computational Biology Department, Carnegie Mellon University, Pittsburgh, 5000 Forbes Ave., PA, USA.
Genome Biol. 2018 Apr 12;19(1):52. doi: 10.1186/s13059-018-1421-5.
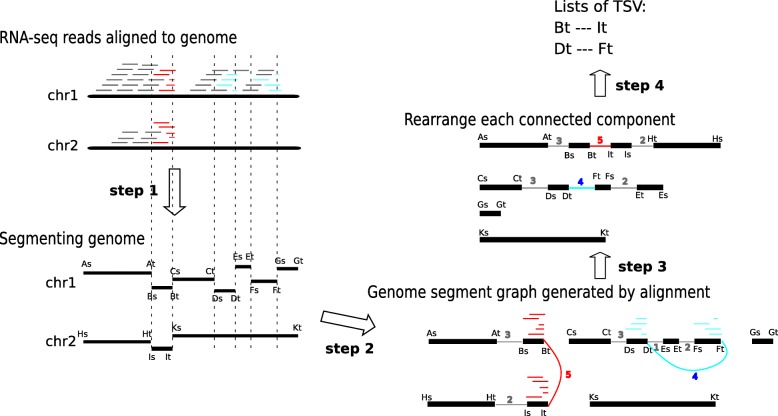
Transcripts are frequently modified by structural variations, which lead to fused transcripts of either multiple genes, known as a fusion gene, or a gene and a previously non-transcribed sequence. Detecting these modifications, called transcriptomic structural variations (TSVs), especially in cancer tumor sequencing, is an important and challenging computational problem. We introduce SQUID, a novel algorithm to predict both fusion-gene and non-fusion-gene TSVs accurately from RNA-seq alignments. SQUID unifies both concordant and discordant read alignments into one model and doubles the precision on simulation data compared to other approaches. Using SQUID, we identify novel non-fusion-gene TSVs on TCGA samples.
转录本经常被结构变异所修饰,这些变异导致多个基因融合的转录本,即融合基因,或者基因和之前不转录的序列融合。检测这些修饰,称为转录组结构变异(TSVs),在癌症肿瘤测序中尤为重要且极具挑战性。我们引入了 SQUID 算法,这是一种新颖的算法,可以从 RNA-seq 比对中准确预测融合基因和非融合基因 TSVs。SQUID 将一致和不一致的读比对统一到一个模型中,与其他方法相比,在模拟数据上的精度提高了一倍。使用 SQUID,我们在 TCGA 样本上发现了新的非融合基因 TSVs。