Institute for Clinical Molecular Biology, University of Kiel, Rosalind-Franklin-Straße 12, 24105, Kiel, Germany.
The Wallenberg Laboratory, Department of Molecular and Clinical Medicine, University of Gothenburg, 41345, Gothenburg, Sweden.
Genome Med. 2018 Apr 13;10(1):27. doi: 10.1186/s13073-018-0534-5.
The interplay of epigenetic processes and the intestinal microbiota may play an important role in intestinal development and homeostasis. Previous studies have established that the microbiota regulates a large proportion of the intestinal epithelial transcriptome in the adult host, but microbial effects on DNA methylation and gene expression during early postnatal development are still poorly understood. Here, we sought to investigate the microbial effects on DNA methylation and the transcriptome of intestinal epithelial cells (IECs) during postnatal development.
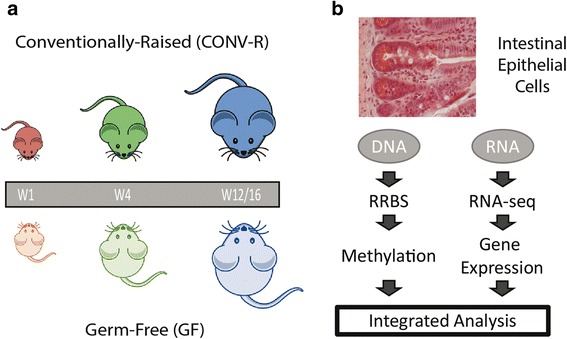
We collected IECs from the small intestine of each of five 1-, 4- and 12 to 16-week-old mice representing the infant, juvenile, and adult states, raised either in the presence or absence of a microbiota. The DNA methylation profile was determined using reduced representation bisulfite sequencing (RRBS) and the epithelial transcriptome by RNA sequencing using paired samples from each individual mouse to analyze the link between microbiota, gene expression, and DNA methylation.
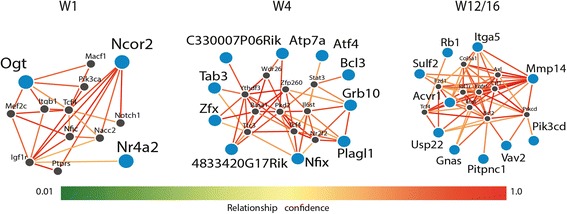
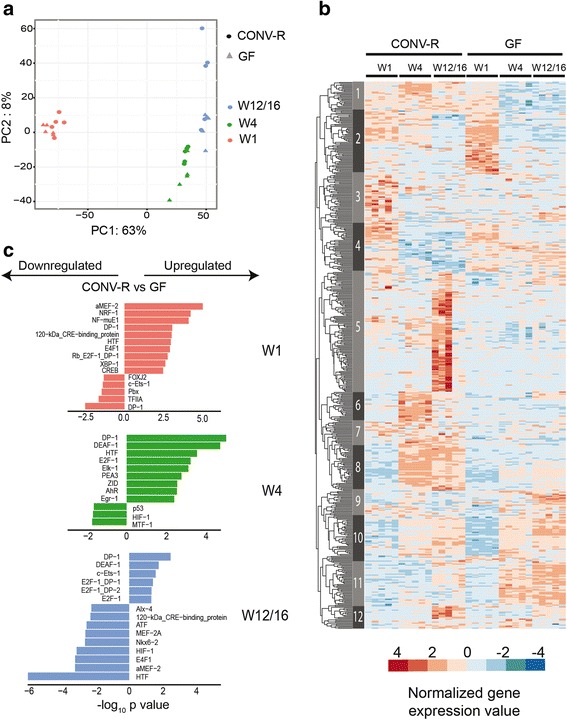
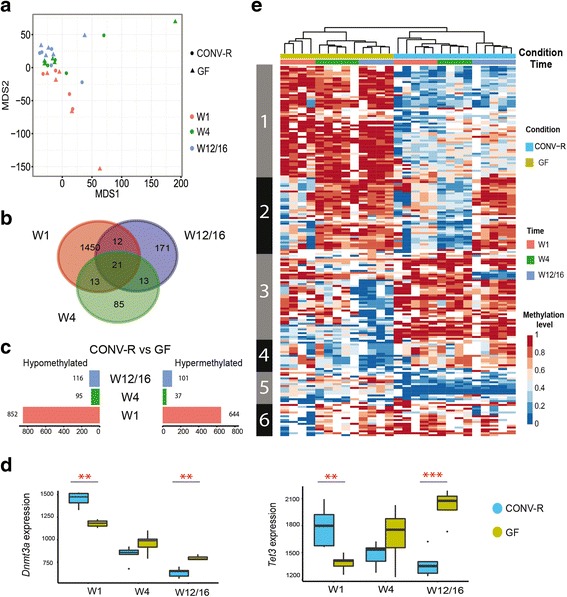
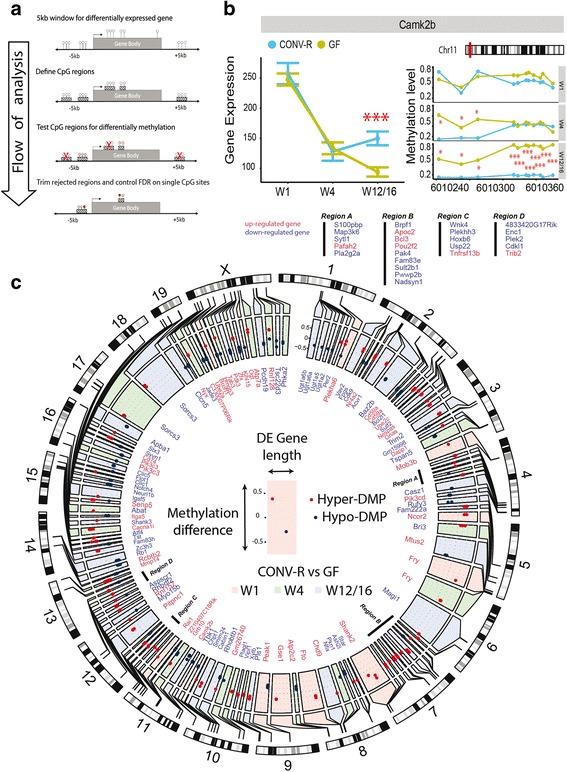
We found that microbiota-dependent and -independent processes act together to shape the postnatal development of the transcriptome and DNA methylation signatures of IECs. The bacterial effect on the transcriptome increased over time, whereas most microbiota-dependent DNA methylation differences were detected already early after birth. Microbiota-responsive transcripts could be attributed to stage-specific cellular programs during postnatal development and regulated gene sets involved primarily immune pathways and metabolic processes. Integrated analysis of the methylome and transcriptome data identified 126 genomic loci at which coupled differential DNA methylation and RNA transcription were associated with the presence of intestinal microbiota. We validated a subset of differentially expressed and methylated genes in an independent mouse cohort, indicating the existence of microbiota-dependent "functional" methylation sites which may impact on long-term gene expression signatures in IECs.
Our study represents the first genome-wide analysis of microbiota-mediated effects on maturation of DNA methylation signatures and the transcriptional program of IECs after birth. It indicates that the gut microbiota dynamically modulates large portions of the epithelial transcriptome during postnatal development, but targets only a subset of microbially responsive genes through their DNA methylation status.
表观遗传过程和肠道微生物群之间的相互作用可能在肠道发育和稳态中发挥重要作用。先前的研究已经确立,微生物群在成年宿主中调节肠道上皮转录组的很大一部分,但微生物对出生后早期发育过程中 DNA 甲基化和基因表达的影响仍知之甚少。在这里,我们试图研究微生物对肠道上皮细胞 (IEC) 出生后发育过程中 DNA 甲基化和转录组的影响。
我们从小肠中收集了 5 只 1、4 和 12 至 16 周龄的小鼠的 IEC,代表婴儿、青少年和成年期,在有或没有微生物群的情况下饲养。使用简化代表性亚硫酸氢盐测序 (RRBS) 测定 DNA 甲基化谱,使用来自每个个体小鼠的配对样本通过 RNA 测序测定上皮转录组,以分析微生物群、基因表达和 DNA 甲基化之间的联系。
我们发现,微生物群依赖和独立的过程共同作用,塑造了 IEC 出生后发育的转录组和 DNA 甲基化特征。细菌对转录组的影响随着时间的推移而增加,而大多数依赖微生物群的 DNA 甲基化差异早在出生后就被检测到。微生物群反应性转录本可以归因于出生后发育过程中的特定阶段特异性细胞程序,并调节主要涉及免疫途径和代谢过程的基因集。甲基组和转录组数据的综合分析确定了 126 个基因组位点,在这些位点上,差异 DNA 甲基化和 RNA 转录与肠道微生物群的存在相关。我们在一个独立的小鼠队列中验证了一组差异表达和甲基化的基因,表明存在依赖微生物群的“功能”甲基化位点,可能影响 IEC 中的长期基因表达特征。
我们的研究代表了第一个对出生后微生物群介导的 DNA 甲基化特征和 IEC 转录程序成熟的全基因组分析。它表明,肠道微生物群在出生后发育过程中动态调节上皮转录组的大部分,但仅通过其 DNA 甲基化状态靶向微生物反应基因的一部分。