Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand.
PLoS One. 2018 Apr 17;13(4):e0195092. doi: 10.1371/journal.pone.0195092. eCollection 2018.
To compare survival of patients with newly diagnosed pulmonary arterial hypertension associated with congenital heart disease (PAH-CHD) according to various clinical classifications with classifications of anatomical-pathophysiological systemic to pulmonary shunts in a single-center cohort.
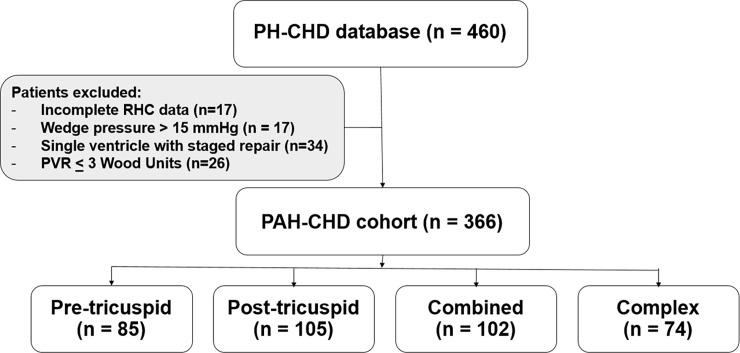
All prevalent cases of PAH-CHD with hemodynamic confirmation by cardiac catheterization in 1995-2015 were retrospectively reviewed. Patients who were younger than three months of age, or with single ventricle following surgery were excluded. Baseline characteristics and clinical outcomes were retrieved from the database. The survival analysis was performed at the end of 2016. Prognostic factors were identified using multivariate analysis.
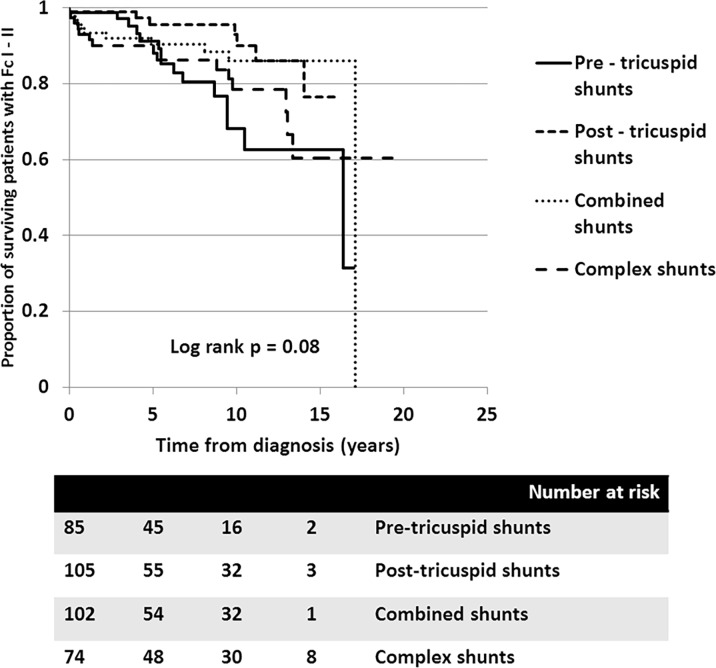
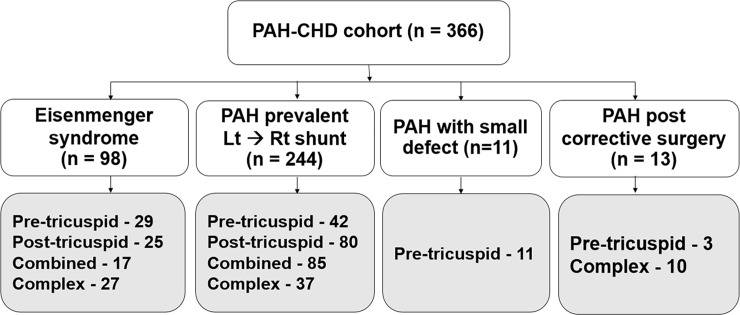
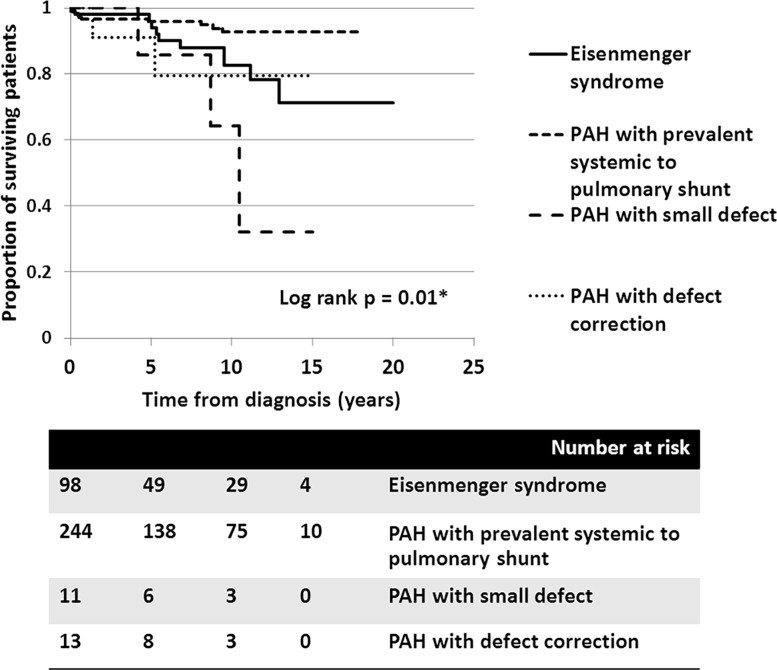
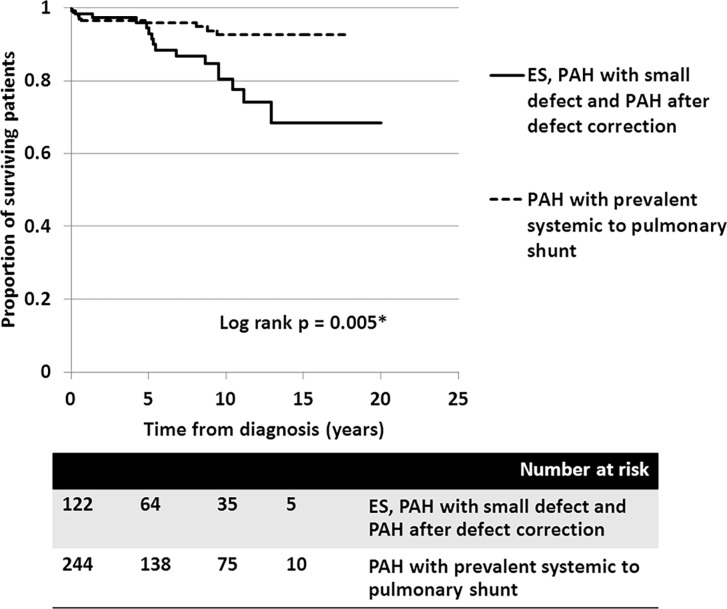
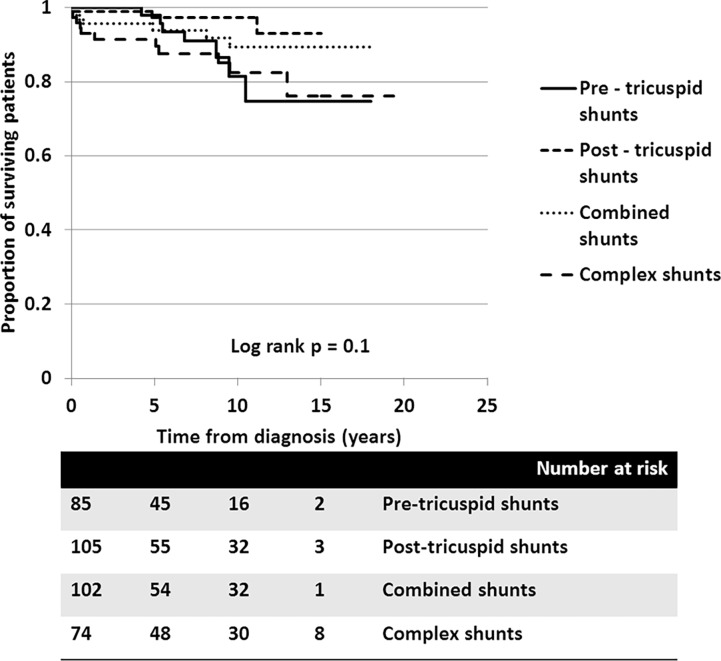
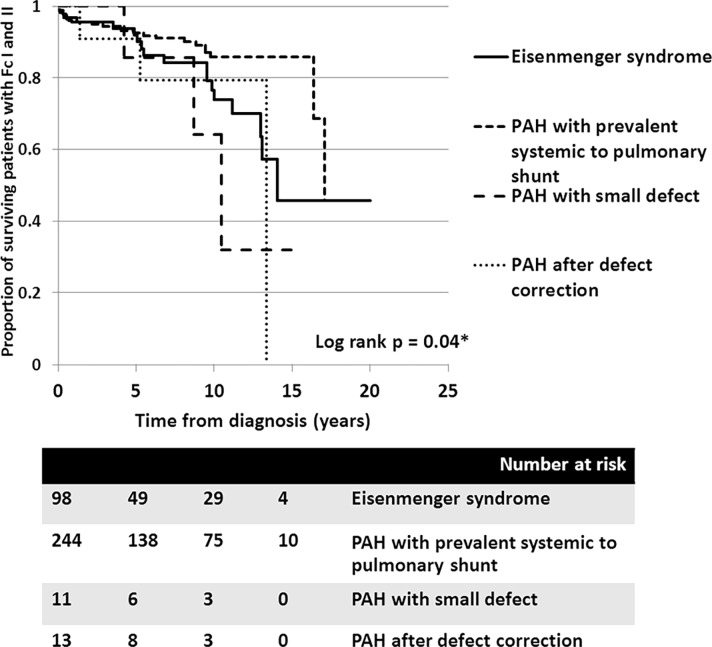
A total of 366 consecutive patients (24.5 ± 17.6 years of age, 40% male) with PAH-CHD were analyzed. Most had simple shunts (85 pre-tricuspid, 105 post-tricuspid, 102 combined shunts). Patients with pre-tricuspid shunts were significantly older at diagnosis in comparison to post-tricuspid, combined, and complex shunts. Clinical classifications identified patients as having Eisenmenger syndrome (ES, 26.8%), prevalent left to right shunt (66.7%), PAH with small defect (3%), or PAH following defect correction (3.5%). At follow-up (median = 5.9 years; 0.1-20.7 years), no statistically significant differences in survival rate were seen among the anatomical-pathophysiological shunts (p = 0.1). Conversely, the clinical classifications revealed that patients with PAH-small defect had inferior survival compared to patients with ES, PAH post-corrective surgery, or PAH with prevalent left to right shunt (p = 0.01). Significant mortality risks were functional class III, age < 10 years, PAH-small defect, elevated right atrial pressure > 15 mmHg, and baseline PVR > 8 WU•m.2.
Patients with PAH-CHD had a modest long-term survival. Different anatomical-pathophysiological shunts affect the natural presentation, while clinical classifications indicate treatment strategies and survival. Contemporary therapy improves survival in deliberately selected patients.
比较单中心队列中,根据各种临床分类和解剖-病理生理系统至肺分流的分类,新诊断的与先天性心脏病相关的肺动脉高压(PAH-CHD)患者的生存情况。
回顾性分析了 1995 年至 2015 年期间经心脏导管检查证实血流动力学为 PAH-CHD 的所有现患病例。排除年龄小于 3 个月或手术后单心室的患者。从数据库中检索基线特征和临床结局。生存分析于 2016 年底进行。使用多变量分析确定预后因素。
共分析了 366 例连续的 PAH-CHD 患者(24.5±17.6 岁,40%为男性)。大多数为单纯分流(85 例三尖瓣前分流,105 例三尖瓣后分流,102 例混合分流)。与三尖瓣后分流、混合分流和复杂分流相比,三尖瓣前分流的患者在诊断时年龄明显更大。临床分类将患者分为艾森曼格综合征(ES,26.8%)、常见左向右分流(66.7%)、肺动脉高压伴小缺损(3%)或肺动脉高压后矫正手术(3.5%)。在随访(中位数=5.9 年;0.1-20.7 年)期间,解剖-病理生理分流之间的生存率无统计学差异(p=0.1)。相反,临床分类显示,与 ES、PAH 矫正手术后或 PAH 常见左向右分流患者相比,PAH 小缺损患者的生存率较低(p=0.01)。显著的死亡风险因素为心功能 III 级、年龄<10 岁、PAH 小缺损、右心房压升高>15mmHg 和基线 PVR>8WU•m.2。
PAH-CHD 患者的长期生存情况尚可。不同的解剖-病理生理分流影响其自然表现,而临床分类则提示治疗策略和生存情况。在经过精心选择的患者中,当代治疗可提高生存率。