Molecular and Cellular Immunology Section, Great Ormond Street Institute of Child Health, University College London, London, United Kingdom.
Department of Pediatric Immunology, Great Ormond Street Hospital for Children NHS Foundation Trust, London, United Kingdom.
Front Immunol. 2018 Apr 4;9:666. doi: 10.3389/fimmu.2018.00666. eCollection 2018.
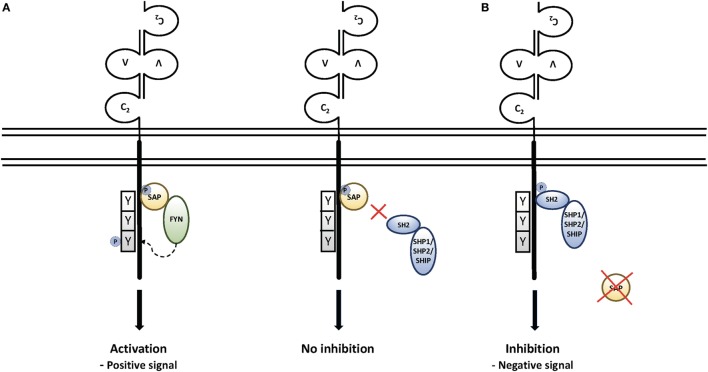
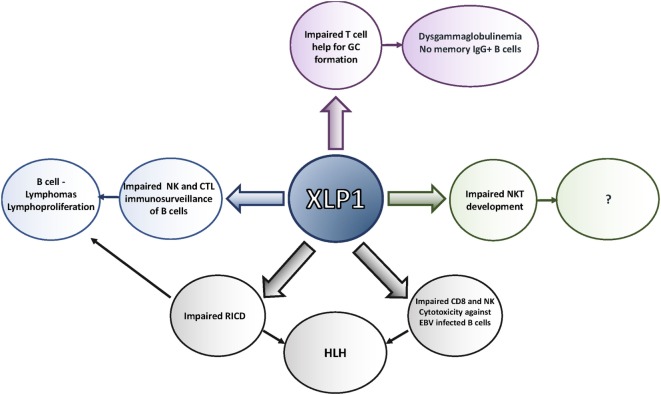
X-linked lymphoproliferative disease (XLP) was first described in the 1970s as a fatal lymphoproliferative syndrome associated with infection with Epstein-Barr virus (EBV). Features include hemophagocytic lymphohistiocytosis (HLH), lymphomas, and dysgammaglobulinemias. Molecular cloning of the causative gene, , has provided insight into the nature of disease, as well as helped characterize multiple features of normal immune cell function. Although XLP type 1 (XLP1) provides an example of a primary immunodeficiency in which patients have problems clearing primarily one infectious agent, it is clear that XLP1 is also a disease of severe immune dysregulation, even independent of EBV infection. Here, we describe clinical features of XLP1, how molecular and biological studies of the gene product, SAP, and the associated signaling lymphocyte activation molecule family receptors have provided insight into disease pathogenesis including specific immune cell defects, and current therapeutic approaches including the potential use of gene therapy. Together, these studies have helped change the outcome of this once almost uniformly fatal disease.
X 连锁淋巴组织增生性疾病(XLP)于 20 世纪 70 年代首次被描述为一种与 EBV 感染相关的致命性淋巴组织增生综合征。其特征包括噬血细胞性淋巴组织细胞增生症(HLH)、淋巴瘤和低丙种球蛋白血症。致病基因的分子克隆,SAP,为疾病的本质提供了深入了解,并有助于描述正常免疫细胞功能的多个特征。尽管 XLP1 型(XLP1)提供了一个原发性免疫缺陷的例子,患者在清除主要传染性病原体方面存在问题,但很明显 XLP1 也是一种严重免疫失调的疾病,甚至与 EBV 感染无关。在这里,我们描述了 XLP1 的临床特征,SAP 的基因产物以及相关信号淋巴细胞激活分子家族受体的分子和生物学研究如何为疾病发病机制提供了深入了解,包括特定免疫细胞缺陷,以及当前的治疗方法,包括基因治疗的潜在应用。这些研究共同帮助改变了这种曾经几乎普遍致命疾病的结局。