Chung Yih-Lin, Wu Mei-Ling
Department of Radiation Oncology, Koo Foundation Sun-Yat-Sen Cancer Center, Taipei 112, Taiwan.
Department of Pathology and Laboratory Medicine, Koo Foundation Sun-Yat-Sen Cancer Center, Taipei 112, Taiwan.
Transl Oncol. 2018 Jun;11(3):743-754. doi: 10.1016/j.tranon.2018.03.013. Epub 2018 Apr 24.
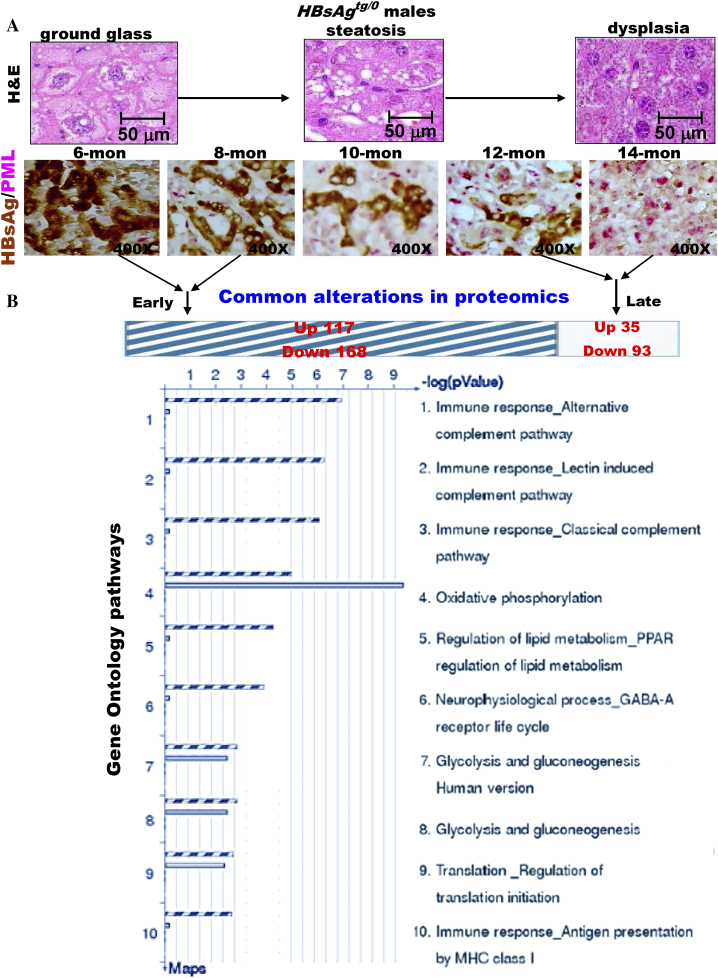
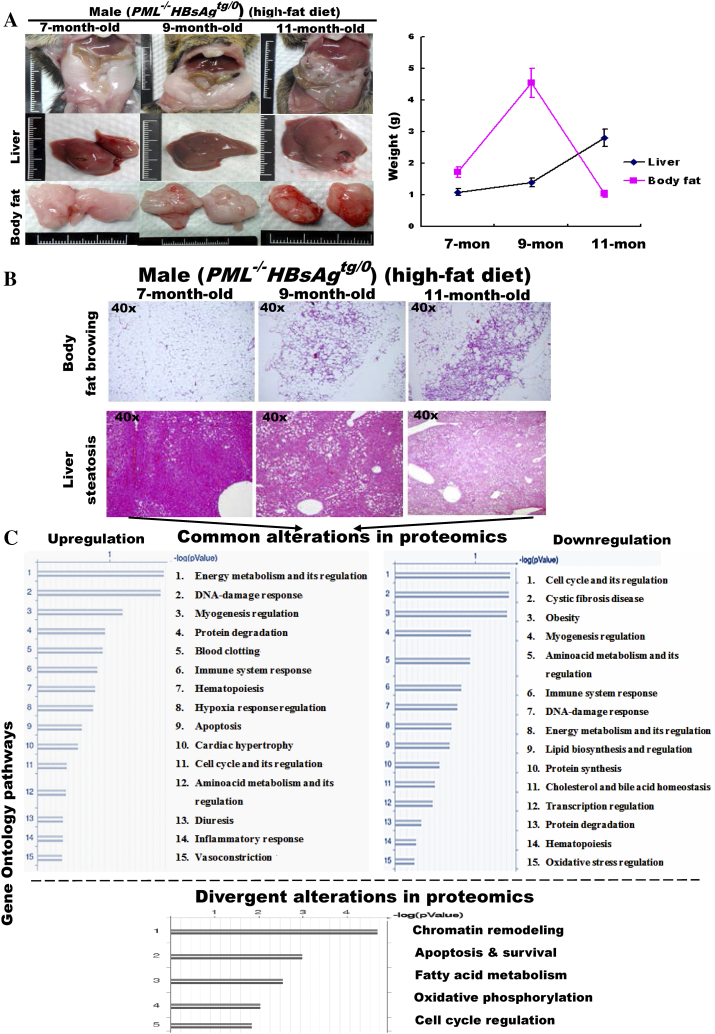
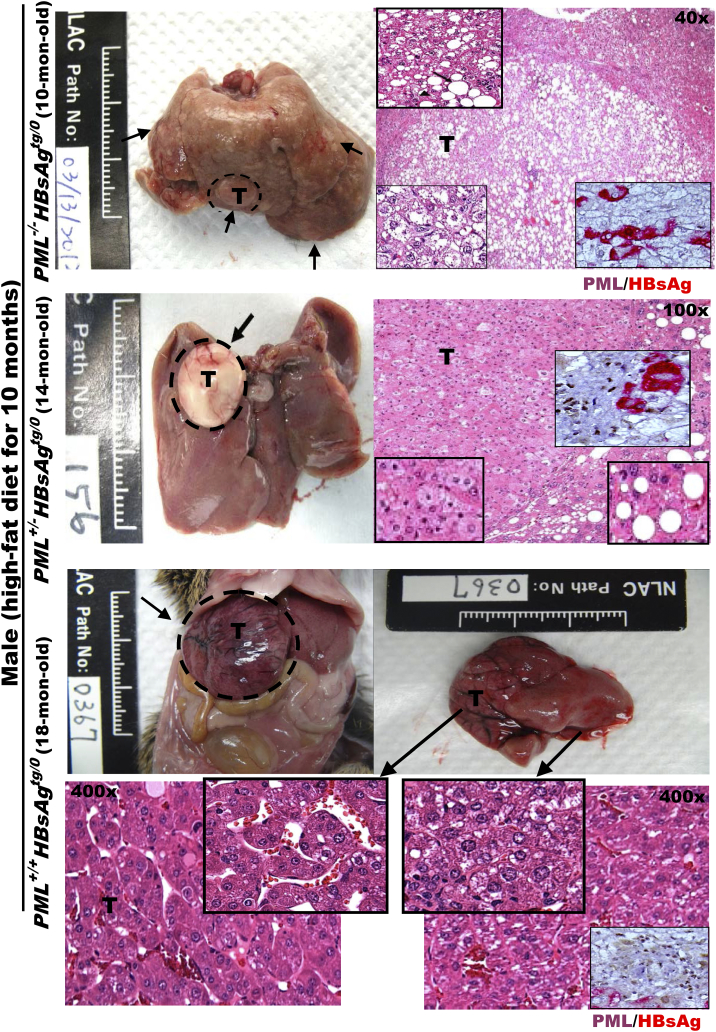
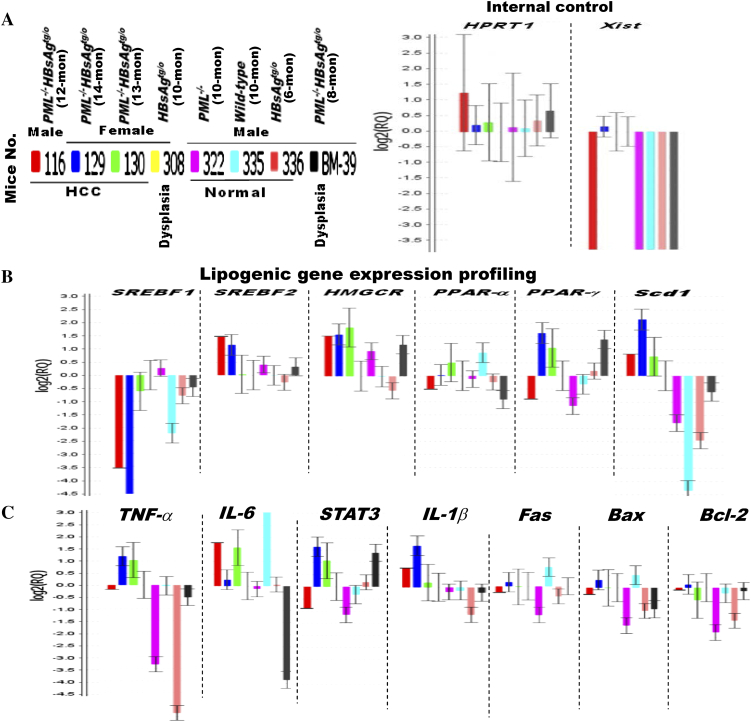
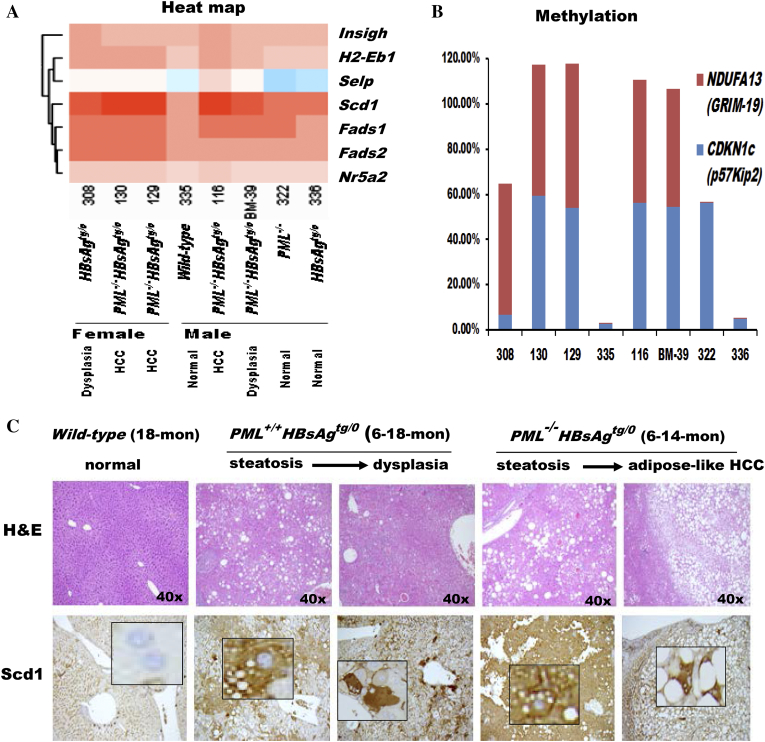
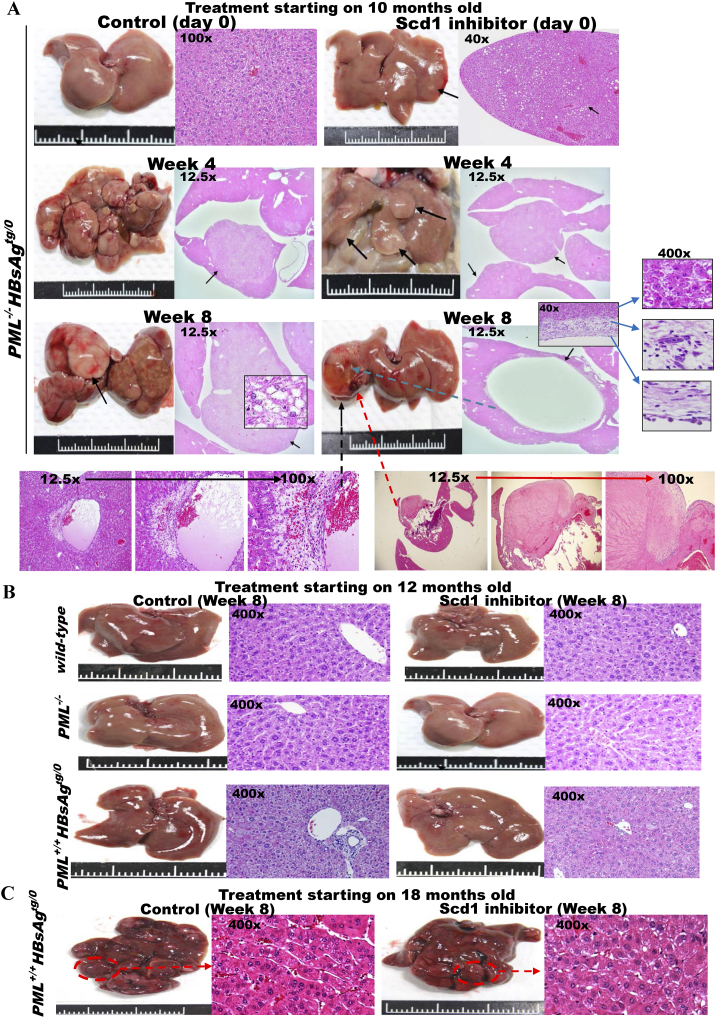
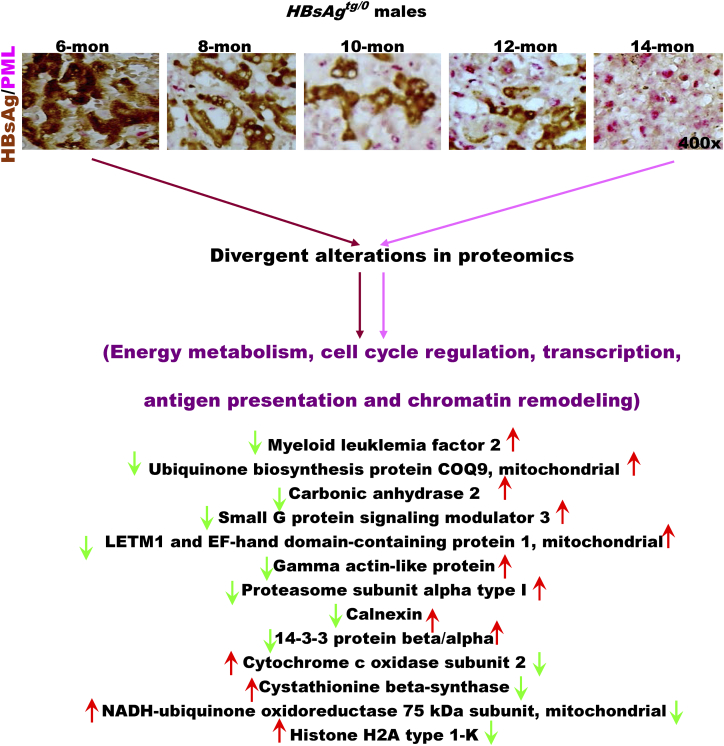
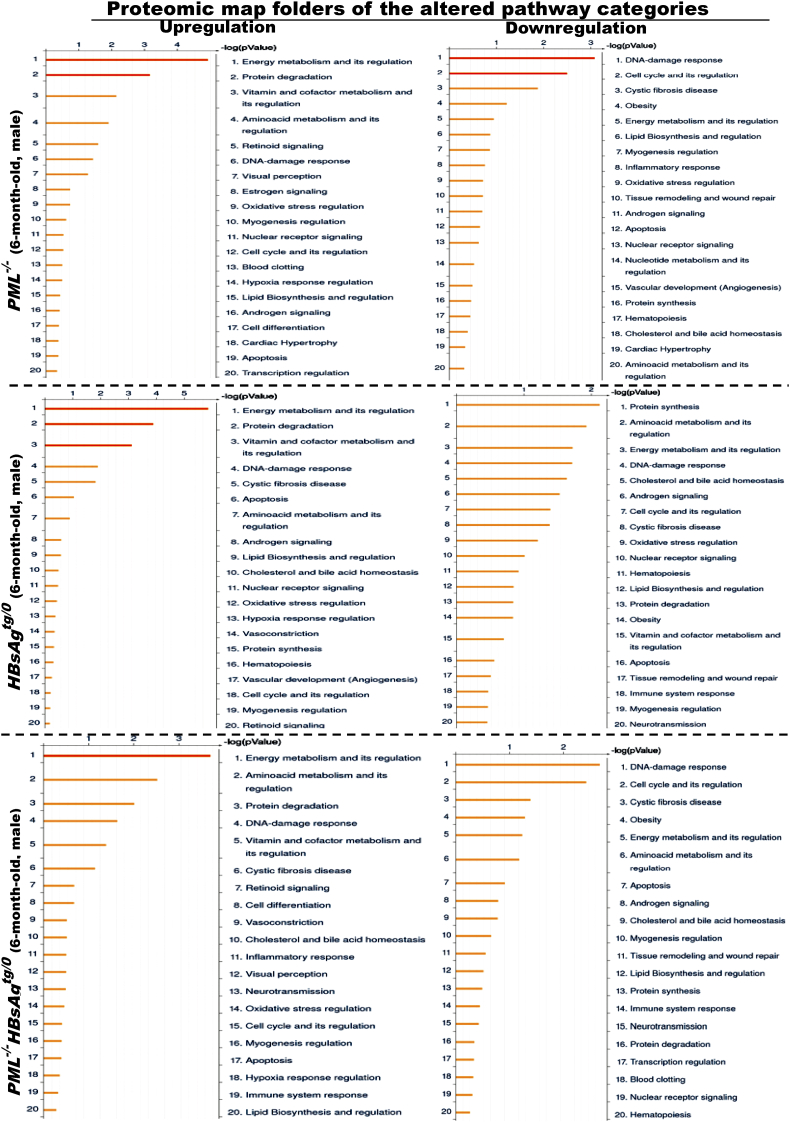
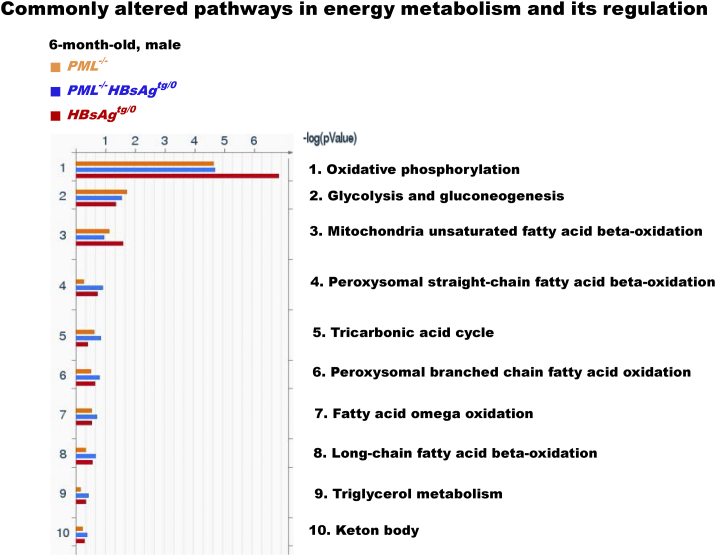
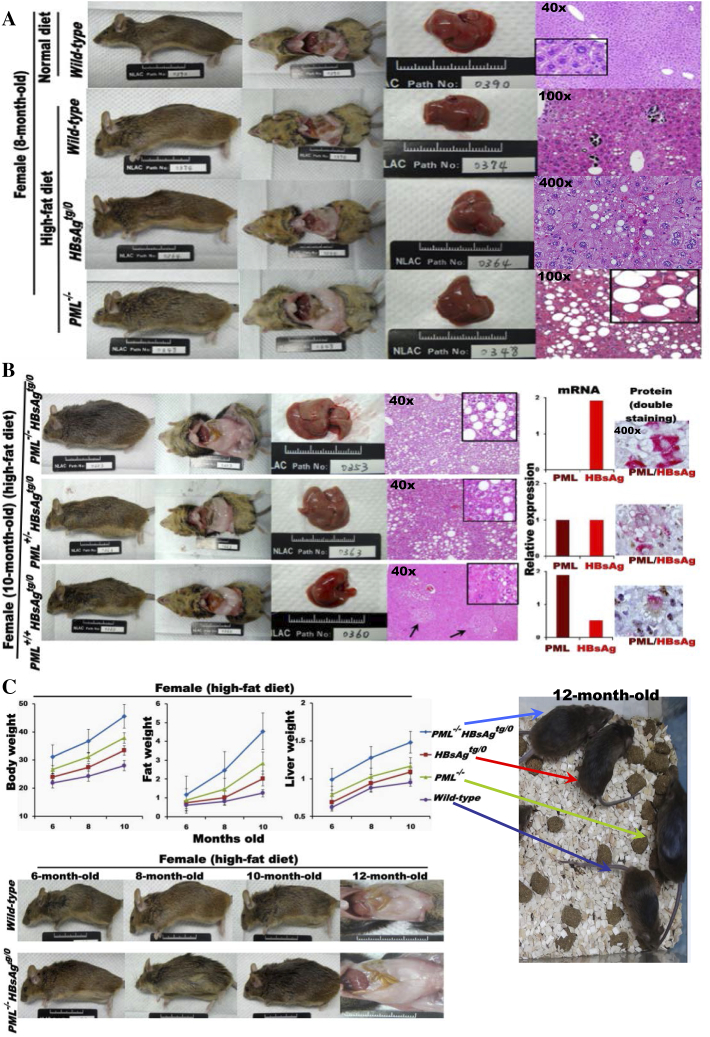
The persistence of hepatitis B surface antigen (HBsAg) is a risk factor for the development of steatosis-associated tumors in chronic hepatitis B virus (HBV) infection, yet little is known about the metabolic link with this factor. We correlated HBV-related pathogenesis in genetically engineered mice and human carriers with metabolic proteomics and lipogenic gene expression profiles. The immunohistochemistry showed that the promyelocytic leukemia protein (PML, a tumor suppressor involved in genome maintenance and fatty acid oxidation), being inversely influenced by the dynamic HBsAg levels from acute phase to seroclearance, appeared as a lipo-metabolic switch linking HBsAg-induced steatosis (lipogenesis) to HBsAg-lost fat-burning hepatocarcinogenesis (lipolysis). Knockdown of PML in HBsAg-transgenic mice predisposed to obesity and drove early steatosis-specific liver tumorigenesis. Proteome analysis revealed that the signaling pathways corresponding to energy metabolism and its regulators were frequently altered by suppression or depletion of PML in the HBsAg-transgenic mice, mainly including oxidative phosphorylation and fatty acid metabolism. Expression profiling further identified upregulation of stearoyl-CoA desaturase 1 (Scd1) and epigenetic methylation of NDUFA13 in the mitochondrial respiratory chain and the cell cycle inhibitor CDKN1c in concordance to the increased severity of lipodystrophy and neoplasia in the livers of HBsAg-transgenic mice with PML insufficiency. The defect in lipolysis in PML-deficient HBsAg-transgenic mice made the HBsAg-induced adipose-like liver tumors vulnerable to synthetic lethality from toxic saturated fat accumulation with a Scd1 inhibitor. Our findings provide mechanistic insights into the evolution of steatosis-associated hepatic tumors driven by reciprocal interactions of HBsAg and PML, and a potential utility of lipid metabolic reprogramming as a treatment target.
乙肝表面抗原(HBsAg)持续存在是慢性乙型肝炎病毒(HBV)感染中脂肪变性相关肿瘤发生的危险因素,但关于该因素与代谢的联系却知之甚少。我们将基因工程小鼠和人类携带者中与HBV相关的发病机制与代谢蛋白质组学和脂肪生成基因表达谱进行了关联分析。免疫组织化学显示,早幼粒细胞白血病蛋白(PML,一种参与基因组维持和脂肪酸氧化的肿瘤抑制因子),受急性期至血清学清除期动态变化的HBsAg水平反向影响,表现为一种脂质代谢开关,将HBsAg诱导的脂肪变性(脂肪生成)与HBsAg消失后的脂肪燃烧性肝癌发生(脂肪分解)联系起来。在易患肥胖症的HBsAg转基因小鼠中敲低PML会引发早期脂肪变性特异性肝肿瘤发生。蛋白质组分析显示,在HBsAg转基因小鼠中,PML的抑制或缺失经常改变与能量代谢及其调节因子相对应的信号通路,主要包括氧化磷酸化和脂肪酸代谢。表达谱分析进一步确定,在PML功能不全的HBsAg转基因小鼠肝脏中,随着脂肪营养不良和肿瘤形成严重程度的增加,硬脂酰辅酶A去饱和酶1(Scd1)上调、线粒体呼吸链中的NDUFA13发生表观遗传甲基化以及细胞周期抑制剂CDKN1c也上调。PML缺陷的HBsAg转基因小鼠脂肪分解缺陷,使得HBsAg诱导的脂肪样肝肿瘤因Scd1抑制剂导致的有毒饱和脂肪积累而容易出现合成致死性。我们的研究结果为HBsAg与PML相互作用驱动的脂肪变性相关肝肿瘤的演变提供了机制性见解,并为脂质代谢重编程作为治疗靶点的潜在应用提供了依据。