Pathology Department, Zhejiang Cancer Hospital, Hangzhou, 3110022, Zhejiang Province, China.
The 2nd Clinical Medical College, Zhejiang Chinese Medical University, Hangzhou, 310053, Zhejiang Province, China.
World J Surg Oncol. 2018 Apr 23;16(1):82. doi: 10.1186/s12957-018-1380-z.
This study aimed to screen sensitive biomarkers for the efficacy evaluation of neoadjuvant chemotherapy in breast cancer.
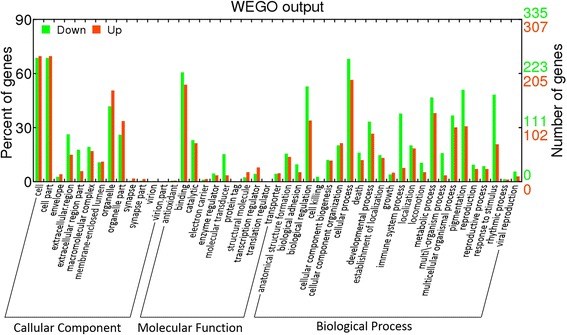
In this study, Illumina digital gene expression sequencing technology was applied and differentially expressed genes (DEGs) between patients presenting pathological complete response (pCR) and non-pathological complete response (NpCR) were identified. Further, gene ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were then performed. The genes in significant enriched pathways were finally quantified by quantitative real-time PCR (qRT-PCR) to confirm that they were differentially expressed. Additionally, GSE23988 from Gene Expression Omnibus database was used as the validation dataset to confirm the DEGs.
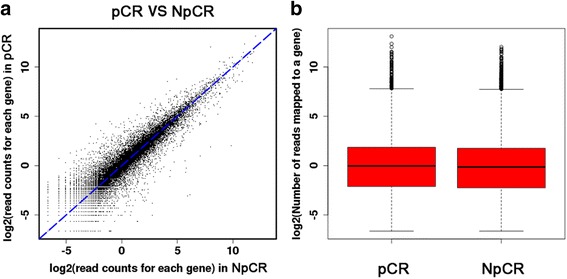
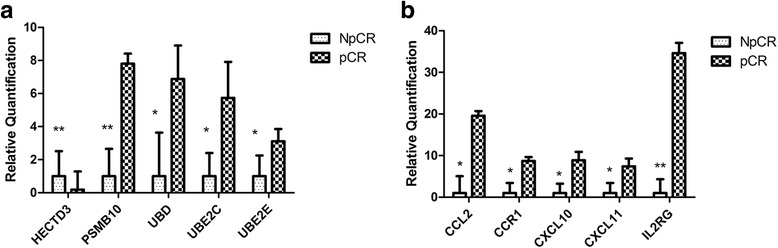
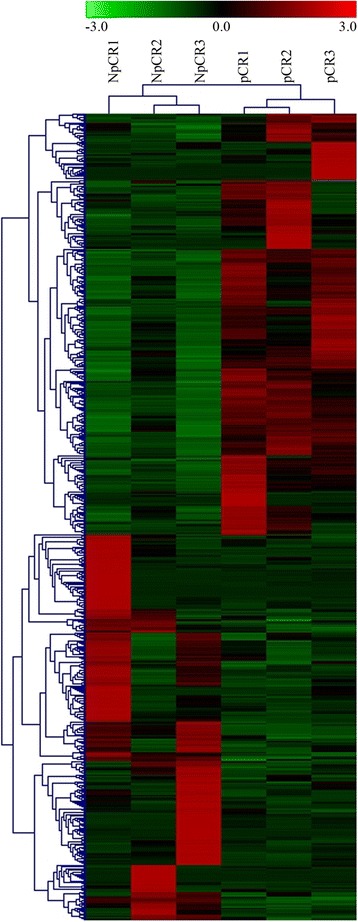
After removing the low-quality reads, 715 DEGs were finally detected. After mapping to KEGG pathways, 10 DEGs belonging to the ubiquitin proteasome pathway (HECTD3, PSMB10, UBD, UBE2C, and UBE2S) and cytokine-cytokine receptor interactions (CCL2, CCR1, CXCL10, CXCL11, and IL2RG) were selected for further analysis. These 10 genes were finally quantified by qRT-PCR to confirm that they were differentially expressed (the log fold changes of selected genes were - 5.34, 7.81, 6.88, 5.74, 3.11, 19.58, 8.73, 8.88, 7.42, and 34.61 for HECTD3, PSMB10, UBD, UBE2C, UBE2S, CCL2, CCR1, CXCL10, CXCL11, and IL2RG, respectively). Moreover, 53 common genes were confirmed by the validation dataset, including downregulated UBE2C and UBE2S.
Our results suggested that these 10 genes belonging to these two pathways might be useful as sensitive biomarkers for the efficacy evaluation of neoadjuvant chemotherapy in breast cancer.
本研究旨在筛选用于评估乳腺癌新辅助化疗疗效的敏感生物标志物。
本研究应用 Illumina 数字基因表达测序技术,鉴定出病理完全缓解(pCR)和非病理完全缓解(NpCR)患者之间的差异表达基因(DEGs)。进一步进行基因本体论和京都基因与基因组百科全书(KEGG)通路富集分析。通过定量实时 PCR(qRT-PCR)对显著富集通路中的基因进行定量,以确认其差异表达。此外,还使用基因表达综合数据库(GEO)中的 GSE23988 数据集作为验证数据集来验证 DEGs。
在去除低质量读数后,最终检测到 715 个 DEGs。在映射到 KEGG 通路后,选择了 10 个属于泛素蛋白酶体途径(HECTD3、PSMB10、UBD、UBE2C 和 UBE2S)和细胞因子-细胞因子受体相互作用(CCL2、CCR1、CXCL10、CXCL11 和 IL2RG)的 DEGs 进行进一步分析。这些 10 个基因最终通过 qRT-PCR 进行定量,以确认其差异表达(所选基因的对数倍变化分别为 HECTD3、PSMB10、UBD、UBE2C、UBE2S、CCL2、CCR1、CXCL10、CXCL11 和 IL2RG 的-5.34、7.81、6.88、5.74、3.11、19.58、8.73、8.88、7.42 和 34.61)。此外,通过验证数据集确认了 53 个共有基因,包括下调的 UBE2C 和 UBE2S。
我们的结果表明,这两个通路中的这 10 个基因可能是用于评估乳腺癌新辅助化疗疗效的敏感生物标志物。