Cell Biology and Neurobiology Branch, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland, United States of America.
Section of Molecular Neurobiology, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland, United States of America.
PLoS Genet. 2018 Apr 26;14(4):e1007363. doi: 10.1371/journal.pgen.1007363. eCollection 2018 Apr.
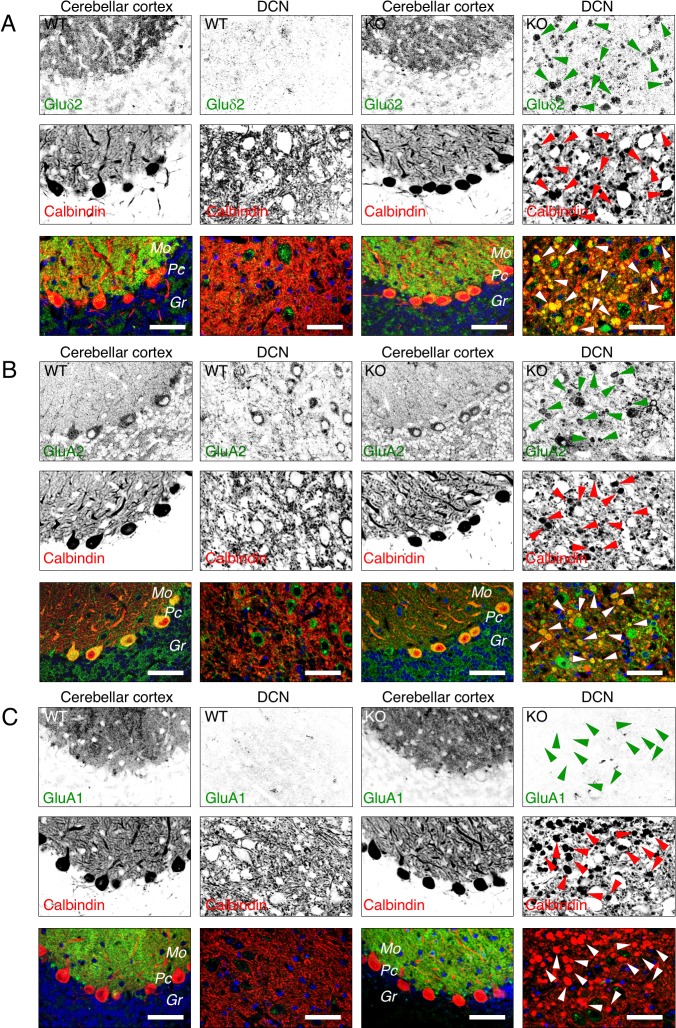
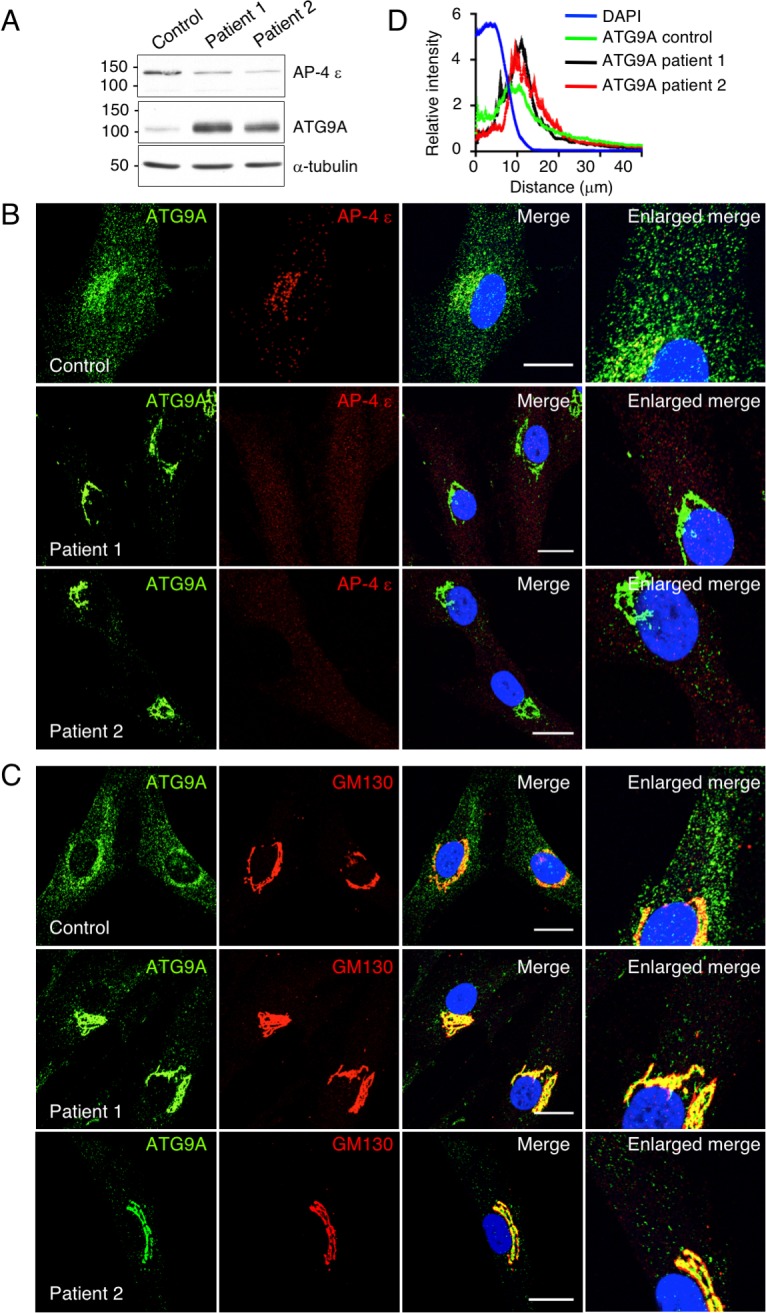
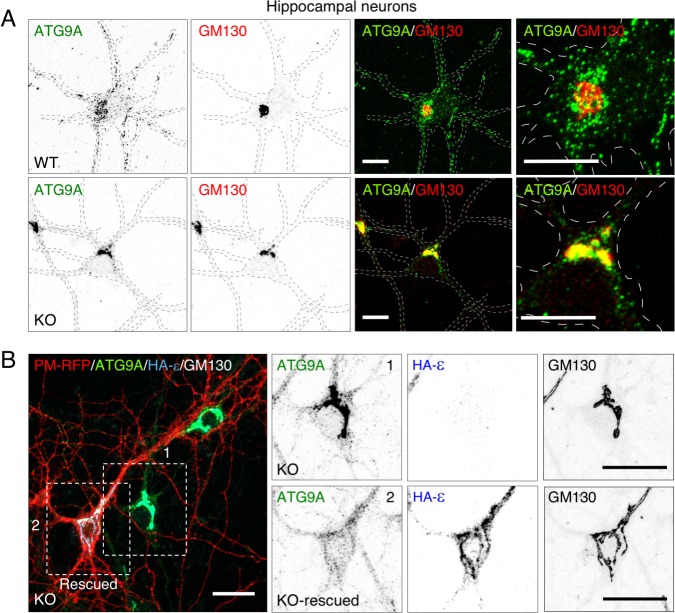
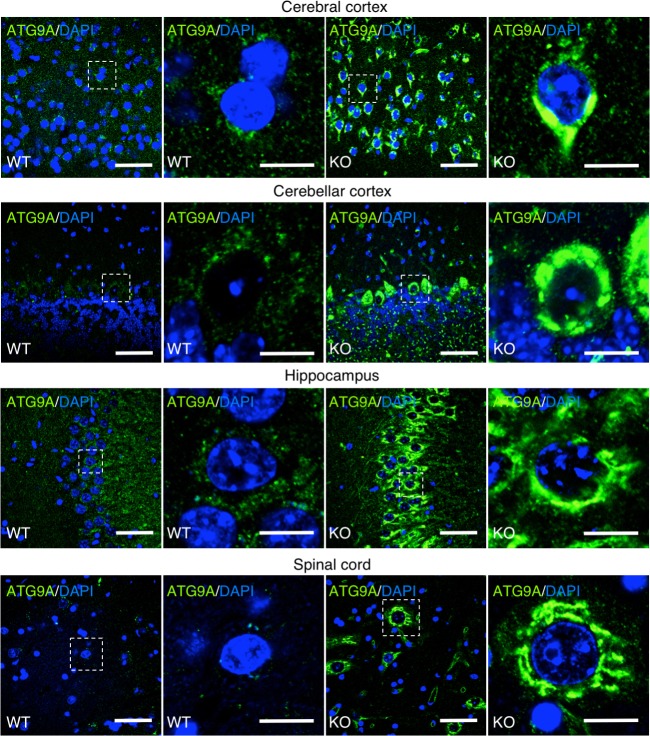
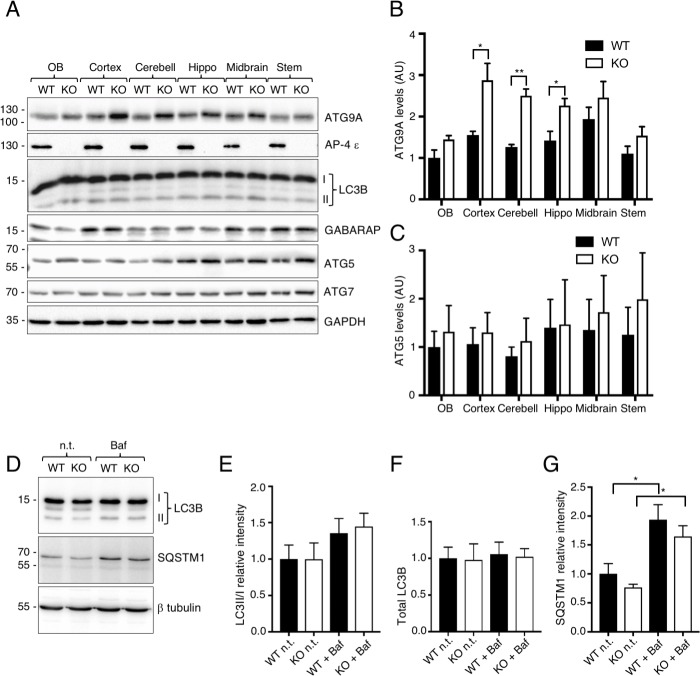
The hereditary spastic paraplegias (HSP) are a clinically and genetically heterogeneous group of disorders characterized by progressive lower limb spasticity. Mutations in subunits of the heterotetrameric (ε-β4-μ4-σ4) adaptor protein 4 (AP-4) complex cause an autosomal recessive form of complicated HSP referred to as "AP-4 deficiency syndrome". In addition to lower limb spasticity, this syndrome features intellectual disability, microcephaly, seizures, thin corpus callosum and upper limb spasticity. The pathogenetic mechanism, however, remains poorly understood. Here we report the characterization of a knockout (KO) mouse for the AP4E1 gene encoding the ε subunit of AP-4. We find that AP-4 ε KO mice exhibit a range of neurological phenotypes, including hindlimb clasping, decreased motor coordination and weak grip strength. In addition, AP-4 ε KO mice display a thin corpus callosum and axonal swellings in various areas of the brain and spinal cord. Immunohistochemical analyses show that the transmembrane autophagy-related protein 9A (ATG9A) is more concentrated in the trans-Golgi network (TGN) and depleted from the peripheral cytoplasm both in skin fibroblasts from patients with mutations in the μ4 subunit of AP-4 and in various neuronal types in AP-4 ε KO mice. ATG9A mislocalization is associated with increased tendency to accumulate mutant huntingtin (HTT) aggregates in the axons of AP-4 ε KO neurons. These findings indicate that the AP-4 ε KO mouse is a suitable animal model for AP-4 deficiency syndrome, and that defective mobilization of ATG9A from the TGN and impaired autophagic degradation of protein aggregates might contribute to neuroaxonal dystrophy in this disorder.
遗传性痉挛性截瘫(HSP)是一组临床表现和遗传异质性的疾病,其特征为进行性下肢痉挛。异四聚体(ε-β4-μ4-σ4)衔接蛋白 4(AP-4)复合物亚基的突变导致常染色体隐性遗传复杂 HSP,称为“AP-4 缺乏综合征”。除了下肢痉挛外,该综合征还表现为智力障碍、小头畸形、癫痫、胼胝体薄和上肢痉挛。然而,其发病机制仍知之甚少。在这里,我们报道了编码 AP-4 的ε 亚基的 AP4E1 基因敲除(KO)小鼠的特征。我们发现,AP-4 ε KO 小鼠表现出一系列神经表型,包括后肢扣状、运动协调能力下降和握力减弱。此外,AP-4 ε KO 小鼠表现出胼胝体变薄和大脑和脊髓各区域的轴突肿胀。免疫组织化学分析表明,跨膜自噬相关蛋白 9A(ATG9A)在皮肤成纤维细胞中在突变μ4 亚基的 AP-4 和各种神经元类型中在 AP-4 ε KO 小鼠中在 TGN 中更集中,并从周围细胞质中耗尽。ATG9A 定位错误与增加的倾向有关,即在 AP-4 ε KO 神经元的轴突中积累突变亨廷顿蛋白(HTT)聚集体。这些发现表明,AP-4 ε KO 小鼠是 AP-4 缺乏综合征的合适动物模型,并且 ATG9A 从 TGN 的动员缺陷和蛋白聚集体的自噬降解受损可能导致该疾病的神经轴突营养不良。