Institute of Human Genetics, University Hospital Jena, Friedrich-Schiller-University Jena, Jena, Germany.
Core Facility Mass Spectrometry, Institute of Biochemistry and Molecular Biology, Medical Faculty, University of Bonn, Bonn, Germany.
Autophagy. 2021 Nov;17(11):3690-3706. doi: 10.1080/15548627.2021.1891848. Epub 2021 Mar 9.
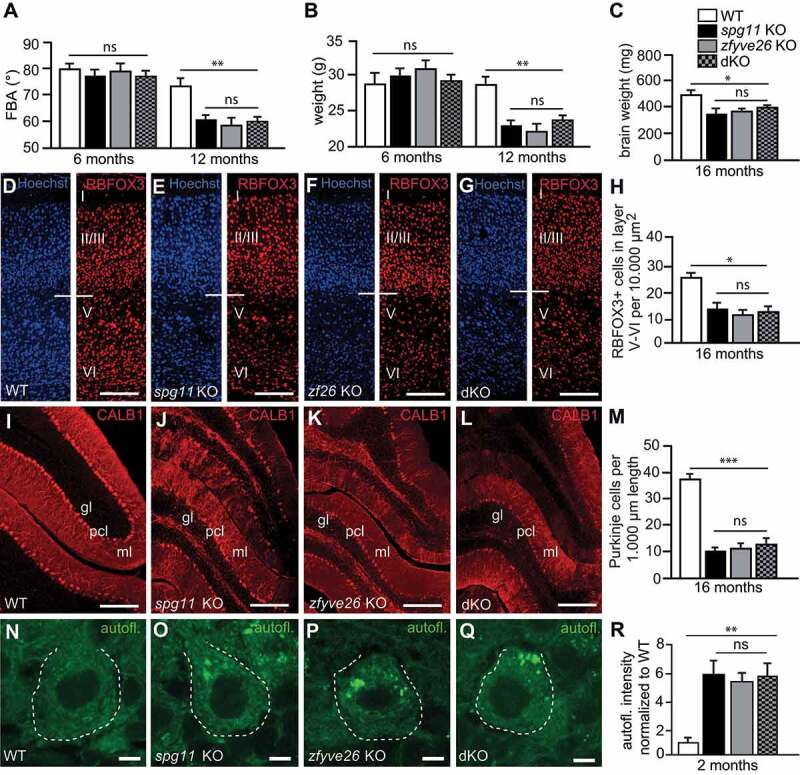
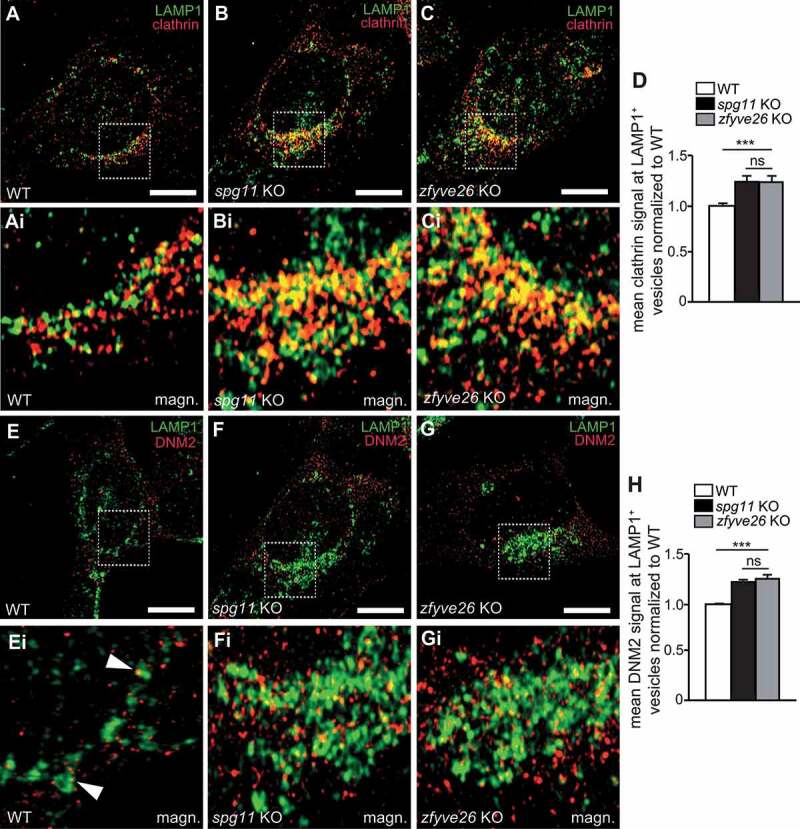
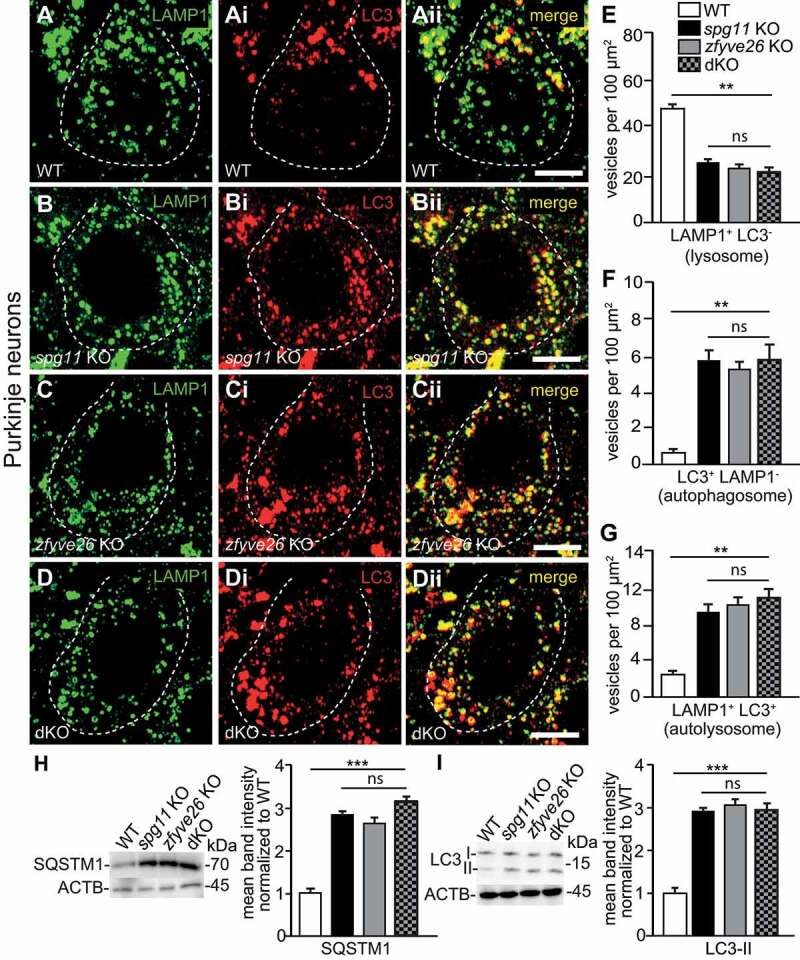
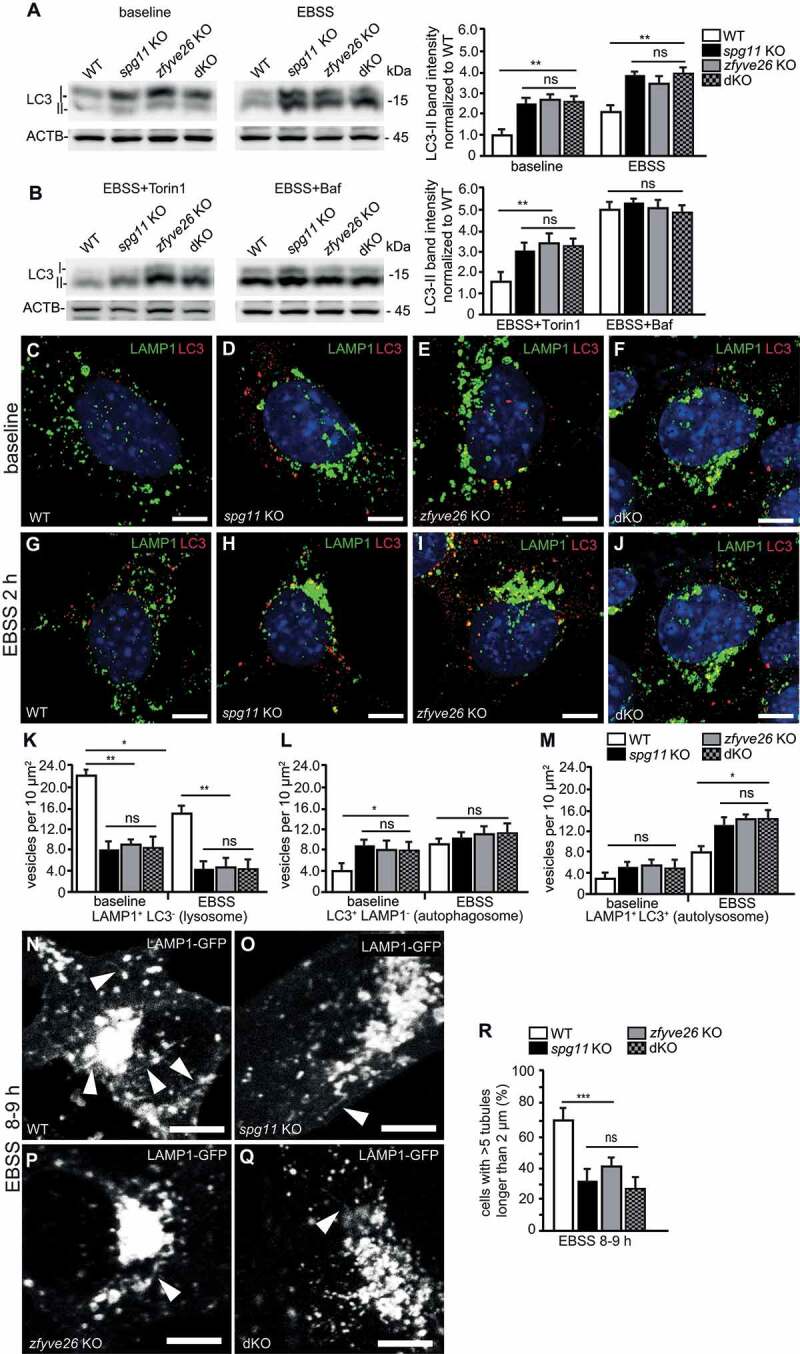
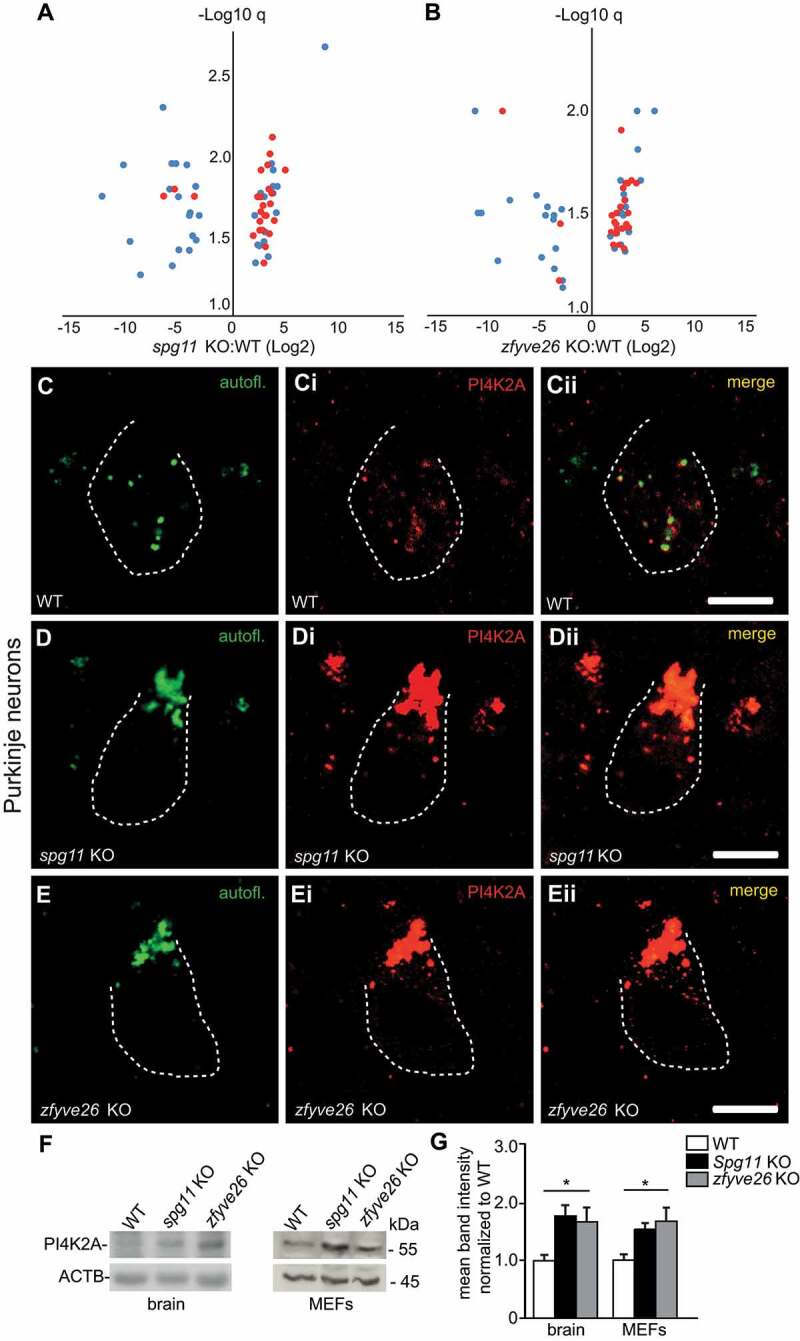
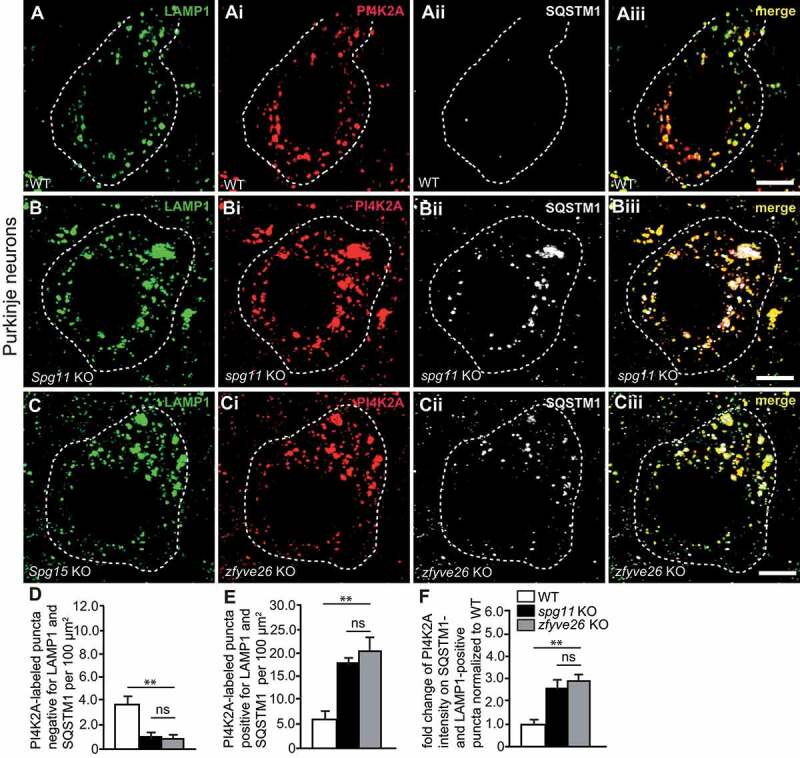
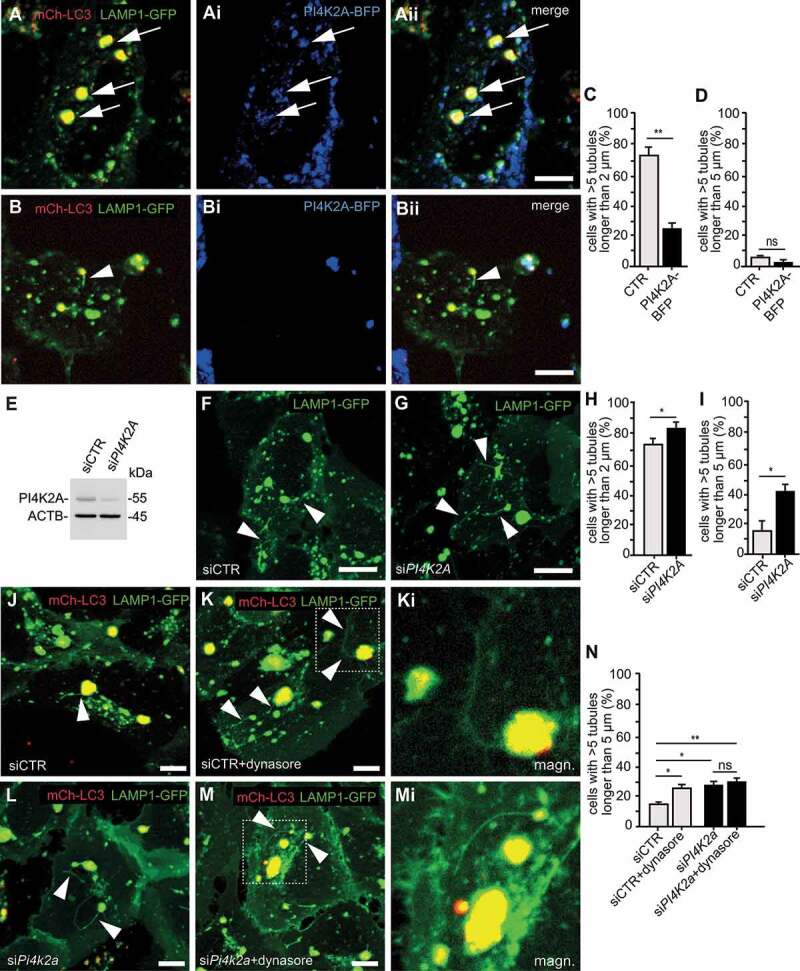
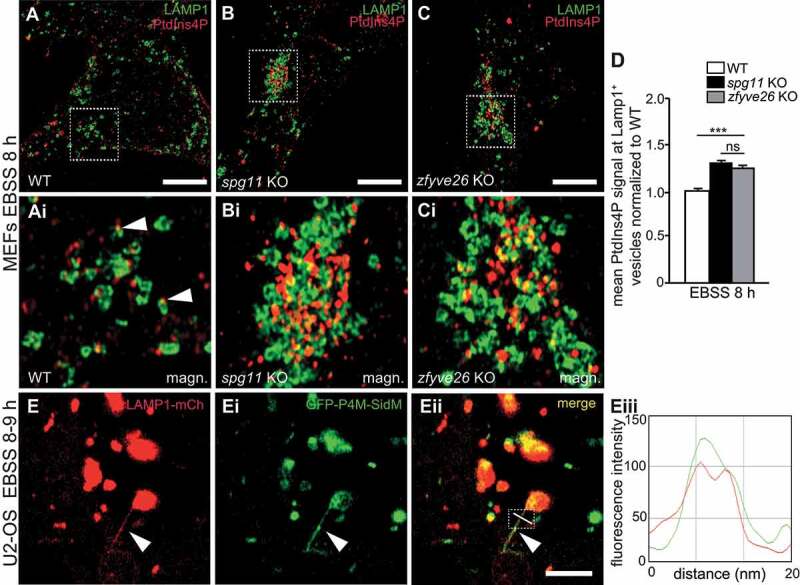
Hereditary spastic paraplegia (HSP) denotes genetically heterogeneous disorders characterized by leg spasticity due to degeneration of corticospinal axons. SPG11 and SPG15 have a similar clinical course and together are the most prevalent autosomal recessive HSP entity. The respective proteins play a role for macroautophagy/autophagy and autophagic lysosome reformation (ALR). Here, we report that and KO mice developed motor impairments within the same course of time. This correlated with enhanced accumulation of autofluorescent material in neurons and progressive neuron loss. In agreement with defective ALR, tubulation events were diminished in starved KO mouse embryonic fibroblasts (MEFs) and lysosomes decreased in neurons of KO brain sections. Confirming that both proteins act in the same molecular pathway, the pathologies were not aggravated upon simultaneous disruption of both. We further show that PI4K2A (phosphatidylinositol 4-kinase type 2 alpha), which phosphorylates phosphatidylinositol to phosphatidylinositol-4-phosphate (PtdIns4P), accumulated in autofluorescent deposits isolated from KO but not WT brains. Elevated PI4K2A abundance was already found at autolysosomes of neurons of presymptomatic KO mice. Immunolabelings further suggested higher levels of PtdIns4P at LAMP1-positive structures in starved KO MEFs. An increased association with LAMP1-positive structures was also observed for clathrin and DNM2/dynamin 2, which are important effectors of ALR recruited by phospholipids. Because PI4K2A overexpression impaired ALR, while its knockdown increased tubulation, we conclude that PI4K2A modulates phosphoinositide levels at autolysosomes and thus the recruitment of downstream effectors of ALR. Therefore, PI4K2A may play an important role in the pathogenesis of SPG11 and SPG15.: ALR: autophagic lysosome reformation; AP-5: adaptor protein complex 5; BFP: blue fluorescent protein; dKO: double knockout; EBSS: Earle's balanced salt solution; FBA: foot base angle; GFP: green fluorescent protein; HSP: hereditary spastic paraplegia; KO: knockout; LAMP1: lysosomal-associated membrane protein 1; MAP1LC3B/LC3: microtubule-associated protein 1 light chain 3 beta; MEF: mouse embryonic fibroblast; SQSTM1/p62: sequestosome 1; PI4K2A: phosphatidylinositol 4-kinase type 2 alpha; PtdIns3P: phosphatidylinositol-3-phosphate; PtdIns4P: phosphatidylinositol-4-phosphate; RFP: red fluorescent protein; SPG: spastic paraplegia gene; TGN: trans-Golgi network; WT: wild type.
遗传性痉挛性截瘫 (HSP) 是指遗传异质性疾病,其特征是由于皮质脊髓束轴突变性导致腿部痉挛。SPG11 和 SPG15 具有相似的临床病程,是最常见的常染色体隐性 HSP 实体。各自的蛋白在巨自噬/自噬和自噬溶酶体再形成 (ALR) 中发挥作用。在这里,我们报告说 和 KO 小鼠在相同的时间内出现运动障碍。这与神经元中自发荧光物质的积累增加和神经元进行性丢失有关。与 ALR 缺陷一致,饥饿的 KO 鼠胚胎成纤维细胞 (MEF) 中的小管化事件减少,KO 脑切片中的溶酶体减少。证实这两种蛋白在同一分子途径中起作用,当同时破坏两者时,病理不会加重。我们进一步表明,PI4K2A(磷脂酰肌醇 4-激酶 2A)在从 KO 而不是 WT 大脑中分离的自发荧光沉积物中积累,PI4K2A 将磷脂酰肌醇磷酸化为磷脂酰肌醇-4-磷酸 (PtdIns4P)。在有症状前 KO 小鼠神经元的自噬溶酶体中已经发现升高的 PI4K2A 丰度。免疫标记进一步表明,在饥饿的 KO MEF 中,LAMP1 阳性结构处的 PtdIns4P 水平更高。在 LAMP1 阳性结构处的结合也观察到网格蛋白和 DNM2/ dynamin 2 的增加,它们是由磷脂募集的 ALR 的重要效应物。由于 PI4K2A 过表达会损害 ALR,而其敲低会增加小管化,因此我们得出结论,PI4K2A 调节自噬溶酶体中的磷酸肌醇水平,从而募集 ALR 的下游效应物。因此,PI4K2A 可能在 SPG11 和 SPG15 的发病机制中发挥重要作用。: ALR: 自噬溶酶体再形成; AP-5: 衔接蛋白复合物 5; BFP: 蓝色荧光蛋白; dKO: 双重敲除; EBSS: Earle 的平衡盐溶液; FBA: 足基部角度; GFP: 绿色荧光蛋白; HSP: 遗传性痉挛性截瘫; KO: 敲除; LAMP1: 溶酶体相关膜蛋白 1; MAP1LC3B/LC3: 微管相关蛋白 1 轻链 3β; MEF: 鼠胚胎成纤维细胞; SQSTM1/p62: 自噬体相关蛋白 1; PI4K2A: 磷脂酰肌醇 4-激酶 2A; PtdIns3P: 磷脂酰肌醇-3-磷酸; PtdIns4P: 磷脂酰肌醇-4-磷酸; RFP: 红色荧光蛋白; SPG: 痉挛性截瘫基因; TGN: 高尔基网络; WT: 野生型。