Department of Otorhinolaryngology, Hearing and Genes, Radboud University Medical Center, Internal Postal Code 377, P.O. Box 9101, 6500 HB, Nijmegen, The Netherlands.
The Radboud Institute for Molecular Life Sciences, Radboud University Medical Center, Nijmegen, The Netherlands.
Hum Genet. 2018 May;137(5):389-400. doi: 10.1007/s00439-018-1880-5. Epub 2018 May 12.
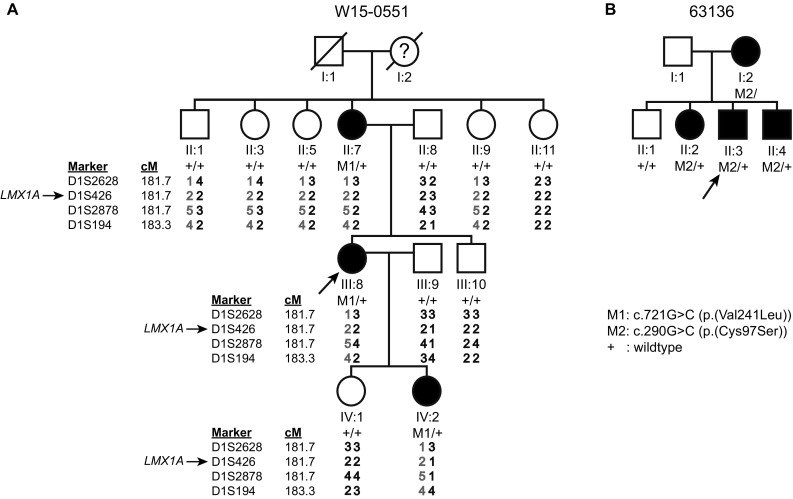
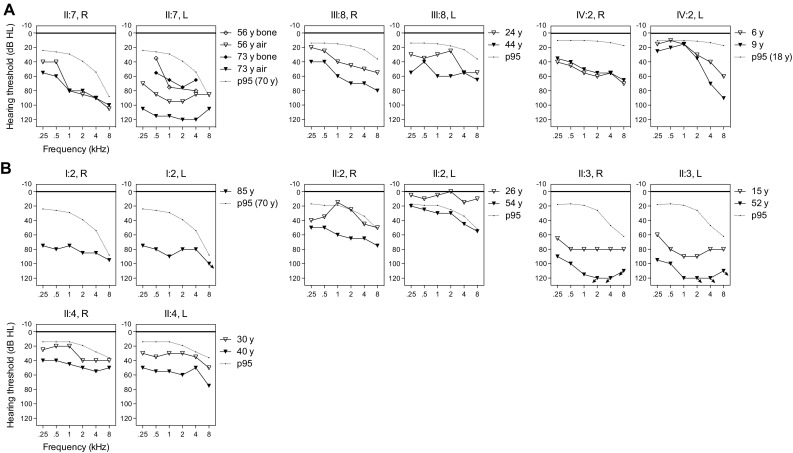
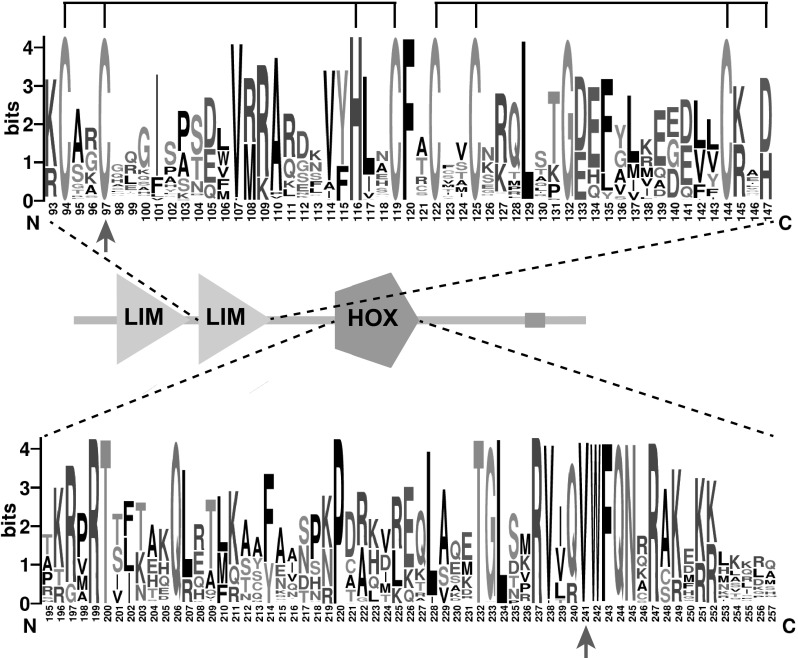
Unraveling the causes and pathomechanisms of progressive disorders is essential for the development of therapeutic strategies. Here, we identified heterozygous pathogenic missense variants of LMX1A in two families of Dutch origin with progressive nonsyndromic hearing impairment (HI), using whole exome sequencing. One variant, c.721G > C (p.Val241Leu), occurred de novo and is predicted to affect the homeodomain of LMX1A, which is essential for DNA binding. The second variant, c.290G > C (p.Cys97Ser), predicted to affect a zinc-binding residue of the second LIM domain that is involved in protein-protein interactions. Bi-allelic deleterious variants of Lmx1a are associated with a complex phenotype in mice, including deafness and vestibular defects, due to arrest of inner ear development. Although Lmx1a mouse mutants demonstrate neurological, skeletal, pigmentation and reproductive system abnormalities, no syndromic features were present in the participating subjects of either family. LMX1A has previously been suggested as a candidate gene for intellectual disability, but our data do not support this, as affected subjects displayed normal cognition. Large variability was observed in the age of onset (a)symmetry, severity and progression rate of HI. About half of the affected individuals displayed vestibular dysfunction and experienced symptoms thereof. The late-onset progressive phenotype and the absence of cochleovestibular malformations on computed tomography scans indicate that heterozygous defects of LMX1A do not result in severe developmental abnormalities in humans. We propose that a single LMX1A wild-type copy is sufficient for normal development but insufficient for maintenance of cochleovestibular function. Alternatively, minor cochleovestibular developmental abnormalities could eventually lead to the progressive phenotype seen in the families.
阐明进行性疾病的病因和发病机制对于治疗策略的制定至关重要。在这里,我们通过全外显子组测序,在两个具有进行性非综合征性听力障碍(HI)的荷兰血统家族中,鉴定出 LMX1A 的杂合致病性错义变异。一个变异,c.721G>C(p.Val241Leu),是新生的,预计会影响 LMX1A 的同源结构域,这对于 DNA 结合至关重要。第二个变异,c.290G>C(p.Cys97Ser),预计会影响第二个 LIM 结构域的一个锌结合残基,该残基参与蛋白质-蛋白质相互作用。Lmx1a 的双等位基因有害变异与小鼠的复杂表型相关,包括耳聋和前庭缺陷,这是由于内耳发育停滞所致。尽管 Lmx1a 小鼠突变体表现出神经系统、骨骼、色素沉着和生殖系统异常,但参与的两个家族的受试者均无综合征特征。此前,LMX1A 被认为是智力障碍的候选基因,但我们的数据不支持这一点,因为受影响的受试者表现出正常的认知能力。HI 的发病年龄(不对称)、严重程度和进展速度存在很大的可变性。大约一半的受影响个体表现出前庭功能障碍,并出现相关症状。迟发性进行性表型以及 CT 扫描未见耳蜗前庭畸形表明,LMX1A 的杂合缺陷不会导致人类出现严重的发育异常。我们提出,单个 LMX1A 野生型拷贝足以促进正常发育,但不足以维持耳蜗前庭功能。或者,耳蜗前庭发育的微小异常最终可能导致家族中所见的进行性表型。