Department of Medicine, Helen Diller Family Comprehensive Cancer Center, University of California, San Francisco, San Francisco, California
Department of Medicine, Helen Diller Family Comprehensive Cancer Center, University of California, San Francisco, San Francisco, California.
Cancer Res. 2018 Jul 1;78(13):3645-3658. doi: 10.1158/0008-5472.CAN-18-0430. Epub 2018 May 14.
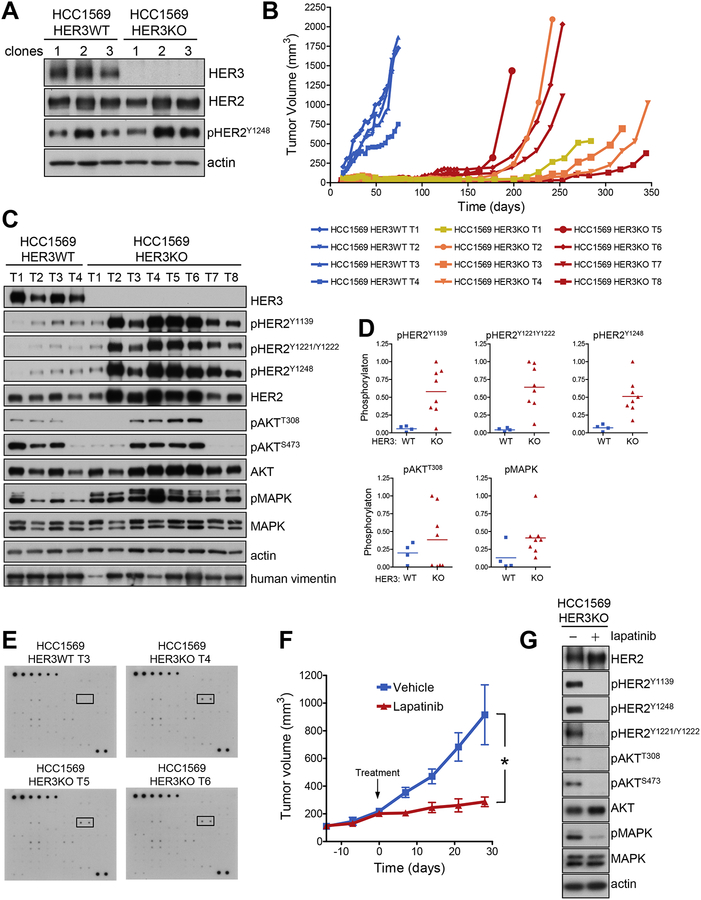
Current evidence suggests that HER2-driven tumorigenesis requires HER3. This is likely due to the unique ability of HER3 to activate PI3K/Akt pathway signaling, which is not directly accessible to HER2. By genetic elimination of HER3 or shRNA knockdown of HER3 in HER2-amplified cancer cells, we find residual HER2-driven activation of PI3K/Akt pathway signaling that is driven by HER2 through direct and indirect mechanisms. Indirect mechanisms involved second messenger pathways, including Ras or Grb2. Direct binding of HER2 to PI3K occurred through p-Tyr1139, which has a weak affinity for PI3K but becomes significant at very high expression and phosphorylation. Mutation of Y1139 impaired the tumorigenic competency of HER2. Total elimination of HER3 expression in HCC1569 HER2-amplified cancer cells significantly impaired tumorigenicity only transiently, overcome by subsequent increases in HER2 expression and phosphorylation with binding and activation of PI3K. In contrast to activation of oncogenes by mutation, activation by overexpression was quantitative in nature: weak intrinsic activities were strengthened by overexpression, with additional gains observed through further increases in expression. Collectively, these data show that progressive functional gains by HER2 can increase its repertoire of activities such as the activation of PI3K and overcome its dependency on HER3. The intrinsic ability of HER2 to activate PI3K correlates with increased HER2 expression and can supplant the dependency upon HER3 for growth in HER2-amplified cancers. .
目前的证据表明,HER2 驱动的肿瘤发生需要 HER3。这可能是由于 HER3 激活 PI3K/Akt 通路信号的独特能力,而 HER2 不能直接激活该通路。通过遗传消除 HER3 或在 HER2 扩增的癌细胞中用 shRNA 敲低 HER3,我们发现残余的 HER2 驱动的 PI3K/Akt 通路信号激活是由 HER2 通过直接和间接机制驱动的。间接机制涉及第二信使途径,包括 Ras 或 Grb2。HER2 与 PI3K 的直接结合发生在 p-Tyr1139 上,它与 PI3K 的亲和力较弱,但在非常高的表达和磷酸化水平下变得显著。Y1139 的突变损害了 HER2 的致瘤能力。在 HCC1569 HER2 扩增癌细胞中完全消除 HER3 表达仅短暂地显著损害了致瘤性,随后 HER2 表达和磷酸化的增加以及 PI3K 的结合和激活克服了这种损害。与突变激活致癌基因不同,过表达激活本质上是定量的:弱固有活性通过过表达得到加强,并通过进一步增加表达获得额外的增益。总之,这些数据表明,HER2 的功能增益可以增加其活性谱,例如激活 PI3K,并克服其对 HER3 的依赖性。HER2 激活 PI3K 的固有能力与 HER2 表达的增加相关,并可以替代 HER2 扩增癌症中对 HER3 的依赖性来促进生长。