Department of Ecology and Evolutionary Biology, University of Michigan, Ann Arbor, Michigan, United States of America.
Department of Animal and Plant Sciences, University of Sheffield, Sheffield, United Kingdom.
PLoS One. 2018 May 17;13(5):e0197433. doi: 10.1371/journal.pone.0197433. eCollection 2018.
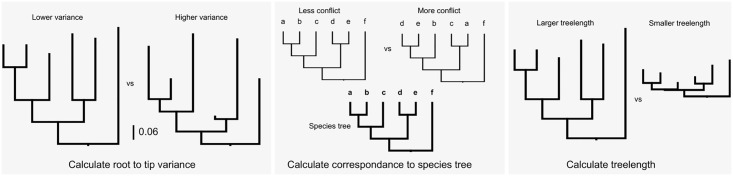
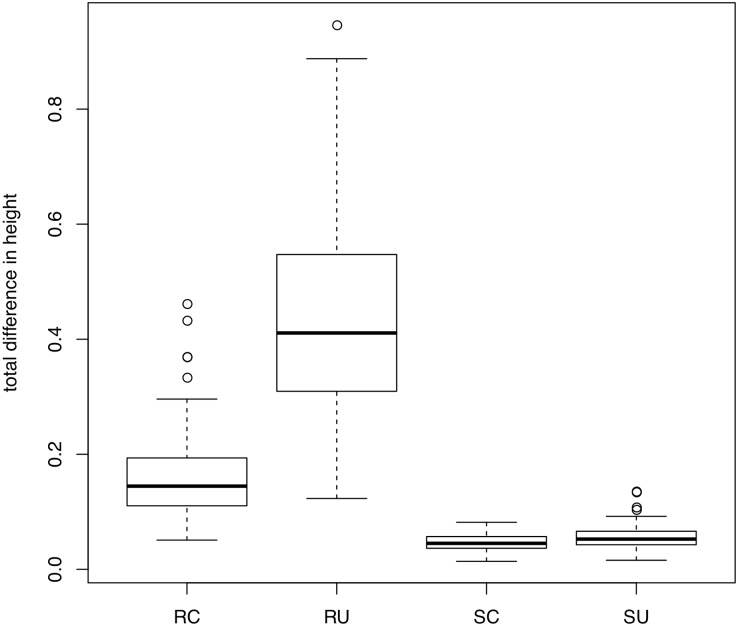
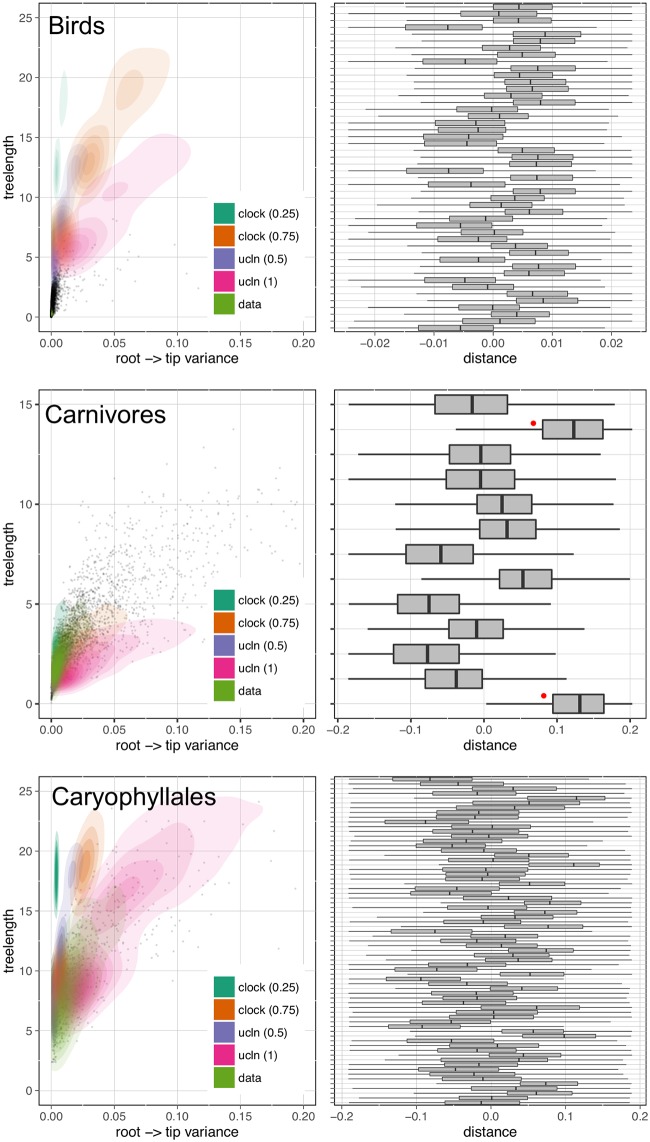
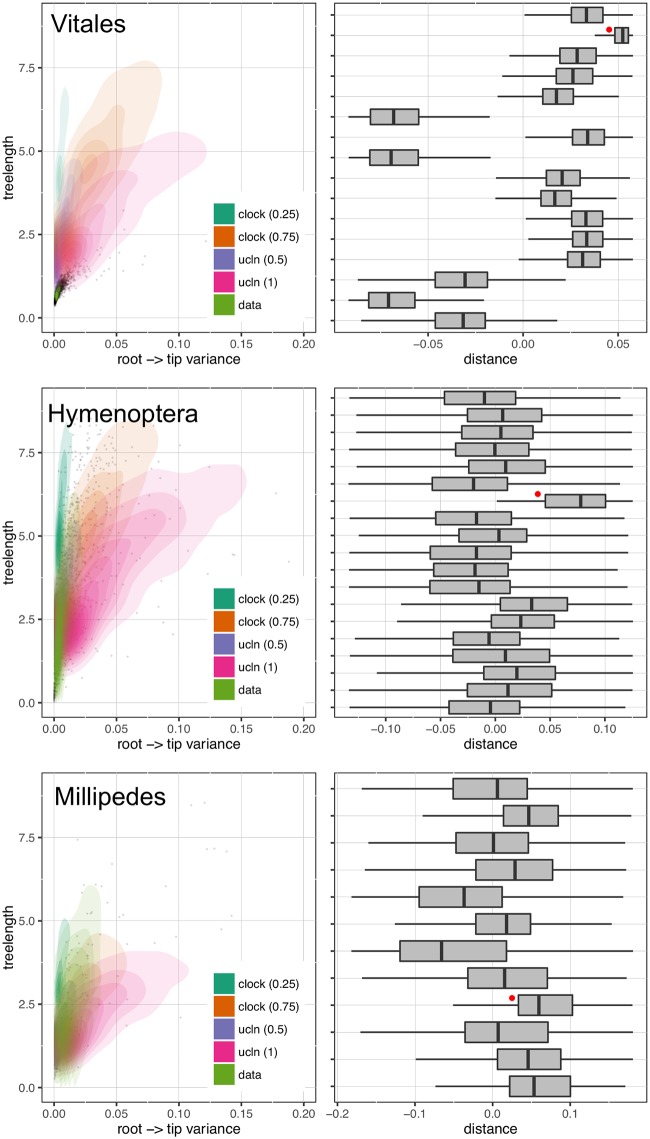
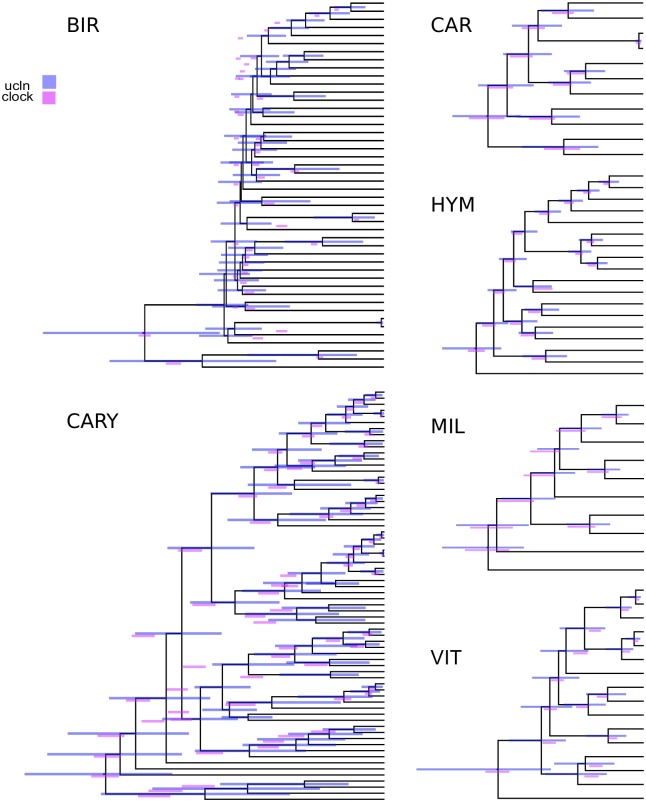
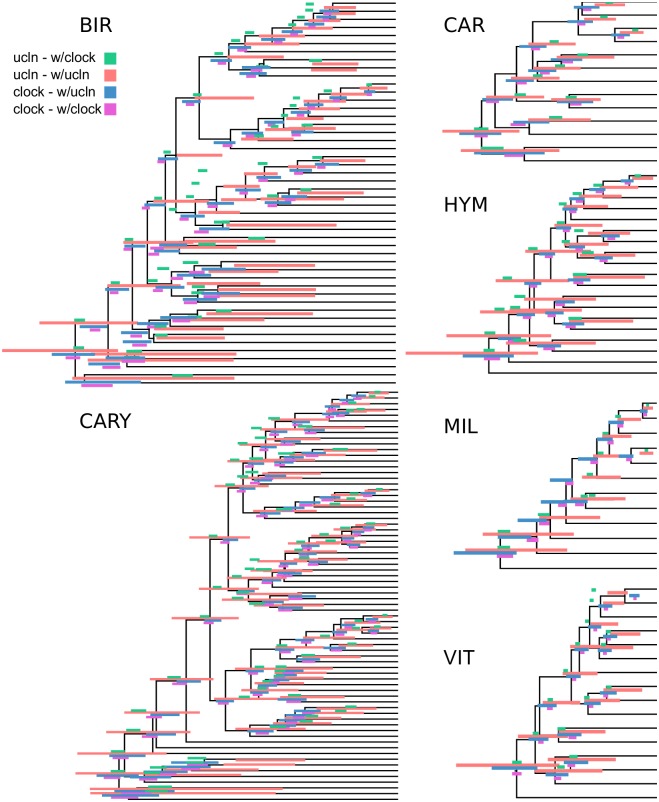
Phylogenomic datasets have been successfully used to address questions involving evolutionary relationships, patterns of genome structure, signatures of selection, and gene and genome duplications. However, despite the recent explosion in genomic and transcriptomic data, the utility of these data sources for efficient divergence-time inference remains unexamined. Phylogenomic datasets pose two distinct problems for divergence-time estimation: (i) the volume of data makes inference of the entire dataset intractable, and (ii) the extent of underlying topological and rate heterogeneity across genes makes model mis-specification a real concern. "Gene shopping", wherein a phylogenomic dataset is winnowed to a set of genes with desirable properties, represents an alternative approach that holds promise in alleviating these issues. We implemented an approach for phylogenomic datasets (available in SortaDate) that filters genes by three criteria: (i) clock-likeness, (ii) reasonable tree length (i.e., discernible information content), and (iii) least topological conflict with a focal species tree (presumed to have already been inferred). Such a winnowing procedure ensures that errors associated with model (both clock and topology) mis-specification are minimized, therefore reducing error in divergence-time estimation. We demonstrated the efficacy of this approach through simulation and applied it to published animal (Aves, Diplopoda, and Hymenoptera) and plant (carnivorous Caryophyllales, broad Caryophyllales, and Vitales) phylogenomic datasets. By quantifying rate heterogeneity across both genes and lineages we found that every empirical dataset examined included genes with clock-like, or nearly clock-like, behavior. Moreover, many datasets had genes that were clock-like, exhibited reasonable evolutionary rates, and were mostly compatible with the species tree. We identified overlap in age estimates when analyzing these filtered genes under strict clock and uncorrelated lognormal (UCLN) models. However, this overlap was often due to imprecise estimates from the UCLN model. We find that "gene shopping" can be an efficient approach to divergence-time inference for phylogenomic datasets that may otherwise be characterized by extensive gene tree heterogeneity.
系统发生基因组数据集已成功用于解决涉及进化关系、基因组结构模式、选择特征以及基因和基因组复制的问题。然而,尽管基因组和转录组数据最近呈爆炸式增长,这些数据源在有效分歧时间推断中的实用性仍未得到检验。系统发生基因组数据集对分歧时间估计提出了两个截然不同的问题:(i)数据量使得整个数据集的推断难以处理,(ii)基因之间潜在的拓扑和速率异质性使得模型误指定成为一个真正的关注点。“基因购物”是一种替代方法,其中系统发生基因组数据集被筛选为一组具有理想特性的基因,这一方法有望缓解这些问题。我们实现了一种针对系统发生基因组数据集的方法(SortaDate 中可用),该方法通过三个标准筛选基因:(i)钟状相似性,(ii)合理的树长(即可分辨的信息含量),以及(iii)与焦点种系树的最小拓扑冲突(假定已经推断出)。这种筛选过程确保了与模型(时钟和拓扑)误指定相关的错误最小化,从而减少了分歧时间估计中的错误。我们通过模拟证明了这种方法的有效性,并将其应用于已发表的动物(鸟类、倍足纲和膜翅目)和植物(肉食性石竹目、广义石竹目和 Vitales)系统发生基因组数据集。通过量化基因和谱系之间的速率异质性,我们发现每个经验数据集都包含具有钟状或几乎钟状行为的基因。此外,许多数据集都有钟状基因,表现出合理的进化速率,并且与种系树大多兼容。在严格的时钟和不相关的对数正态(UCLN)模型下分析这些筛选基因时,我们确定了年龄估计的重叠。然而,这种重叠通常是由于 UCLN 模型的不精确估计造成的。我们发现,对于可能具有广泛基因树异质性的系统发生基因组数据集,“基因购物”可以是一种有效的分歧时间推断方法。