Crespo-Otero Rachel, Kungwan Nawee, Barbatti Mario
School of Biological and Chemical Sciences , Queen Mary University of London , Mile End Road , London E1 4NS , UK . Email:
Department of Chemistry , Faculty of Science , Chiang Mai University , Chiang Mai 50200 , Thailand . Email:
Chem Sci. 2015 Oct 1;6(10):5762-5767. doi: 10.1039/c5sc01902h. Epub 2015 Jul 8.
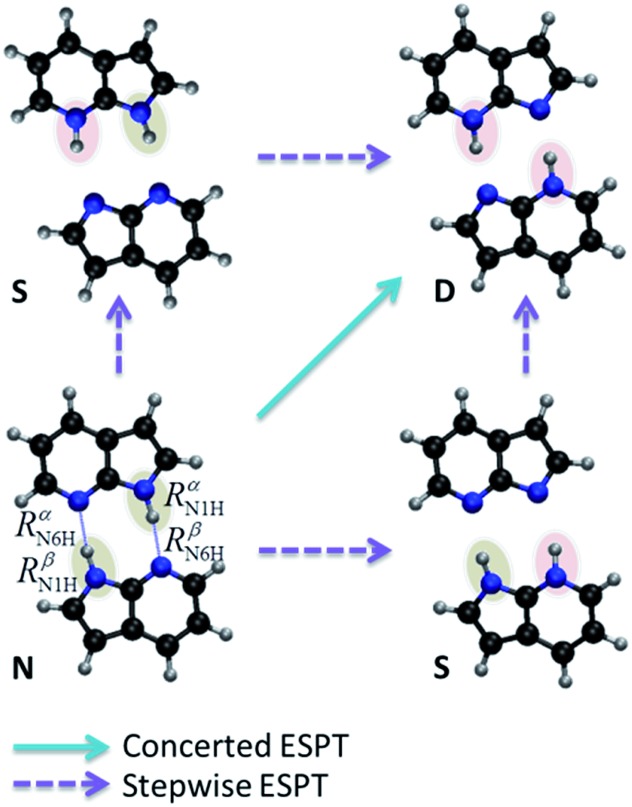
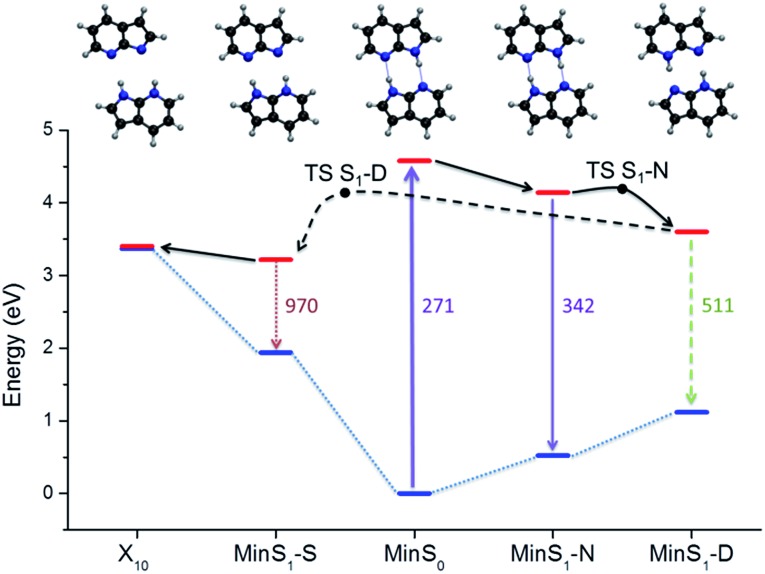
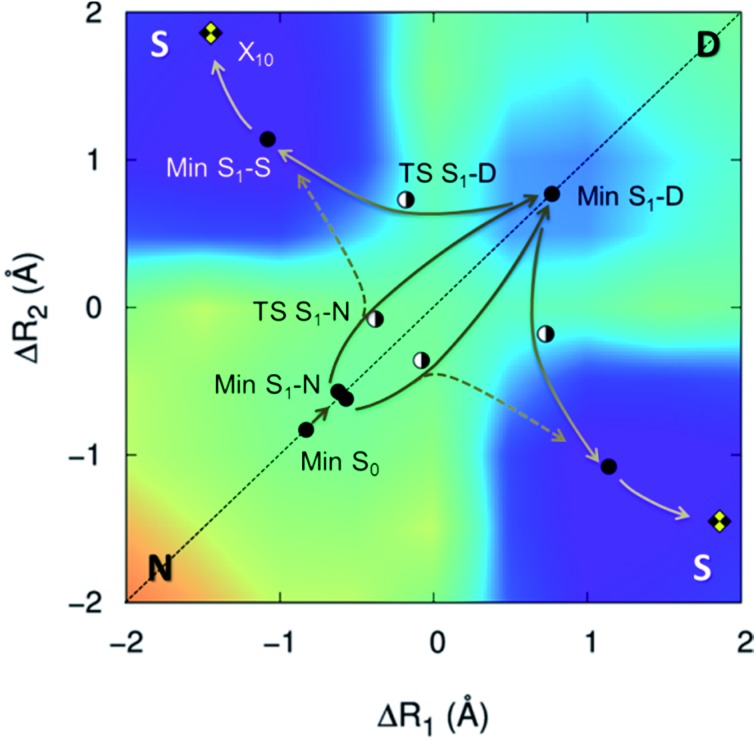
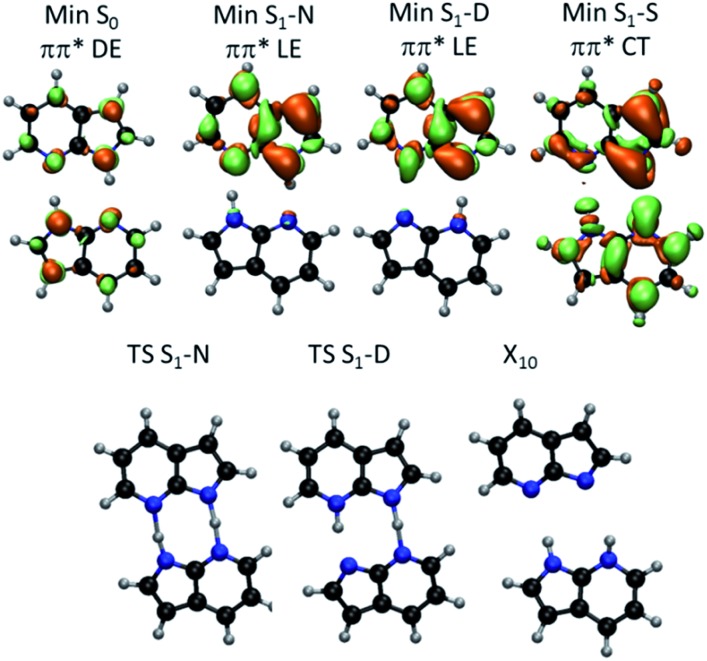
The nature of the excited-state double proton transfer in 7-azaindole (7AI) dimer-whether it is stepwise or concerted-has been under a fierce debate for two decades. Based on high-level computational simulations of static and dynamic properties, we show that much of the earlier discussions was induced by inappropriate theoretical modelling, which led to biased conclusions towards one or other mechanism. A proper topographical description of the excited-state potential energy surface of 7AI dimer in the gas phase clearly reveals that the stepwise mechanism is not accessible due to kinetic and thermodynamic reasons. Single proton transfer can occur, but when it does, an energy barrier blocks the transfer of the second proton and the dimer relaxes through internal conversion. Double proton transfer takes place exclusively by an asynchronous concerted mechanism. This case-study illustrates how computational simulations may lead to unphysical interpretation of experimental results.
二十年来,7-氮杂吲哚(7AI)二聚体中激发态双质子转移的本质——是分步进行还是协同进行——一直处于激烈的争论之中。基于对静态和动态性质的高水平计算模拟,我们表明,许多早期的讨论是由不恰当的理论建模引起的,这导致了对一种或另一种机制的有偏差的结论。对气相中7AI二聚体激发态势能面的恰当拓扑描述清楚地表明,由于动力学和热力学原因,分步机制是不可行的。单质子转移可以发生,但当它发生时,一个能垒会阻止第二个质子的转移,并且二聚体通过内转换弛豫。双质子转移仅通过异步协同机制发生。这个案例研究说明了计算模拟如何可能导致对实验结果的非物理性解释。