Rando Halie M, Farré Marta, Robson Michael P, Won Naomi B, Johnson Jennifer L, Buch Ronak, Bastounes Estelle R, Xiang Xueyan, Feng Shaohong, Liu Shiping, Xiong Zijun, Kim Jaebum, Zhang Guojie, Trut Lyudmila N, Larkin Denis M, Kukekova Anna V
Illinois Informatics Institute, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA.
Department of Animal Science, College of Agricultural, Consumer and Environmental Sciences, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA.
Genes (Basel). 2018 Jun 20;9(6):308. doi: 10.3390/genes9060308.
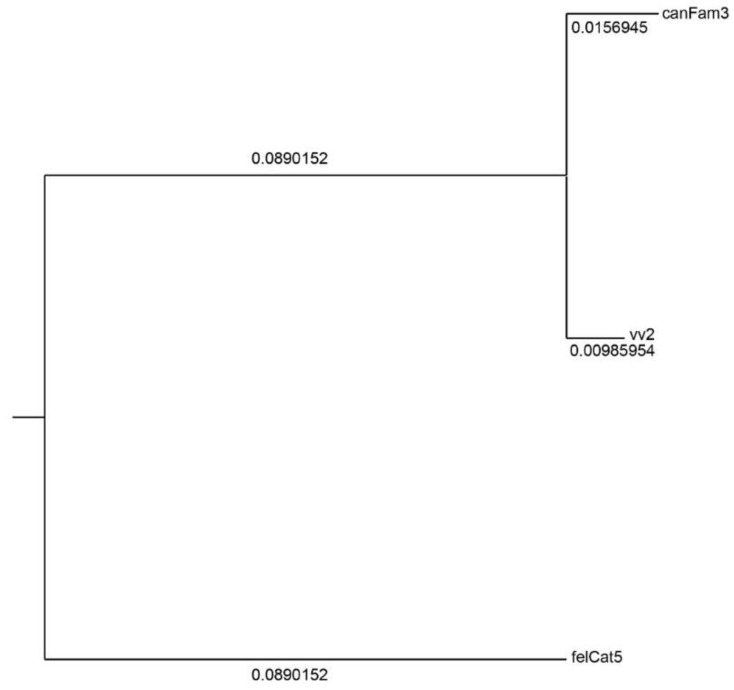
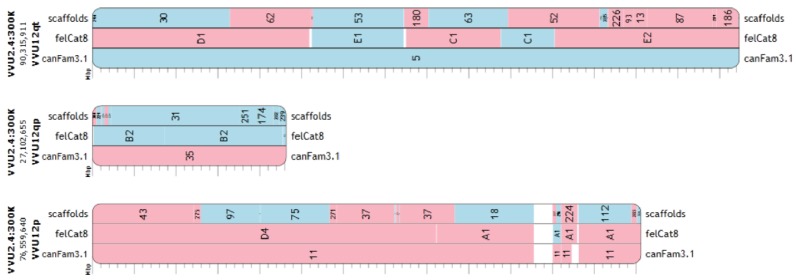
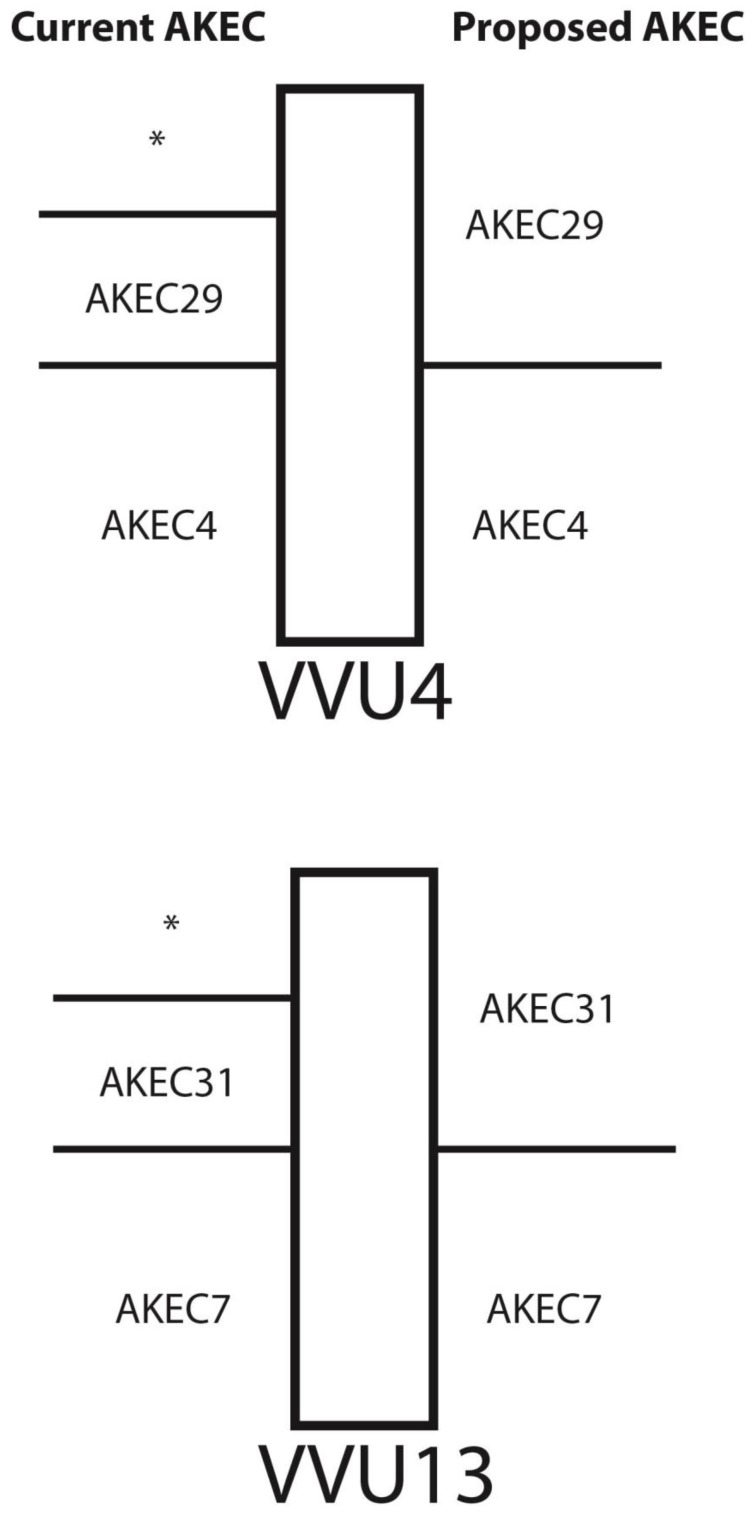
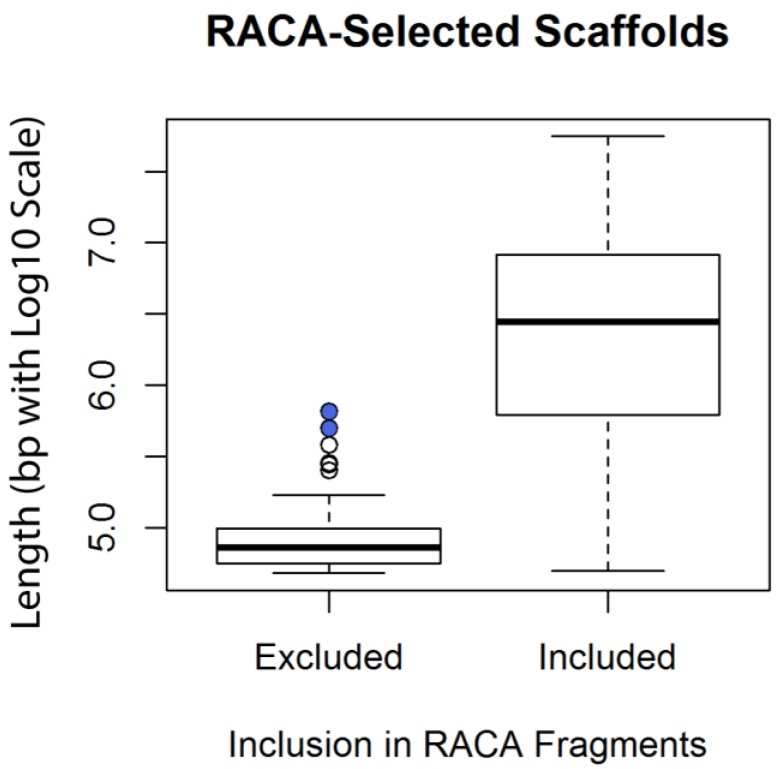
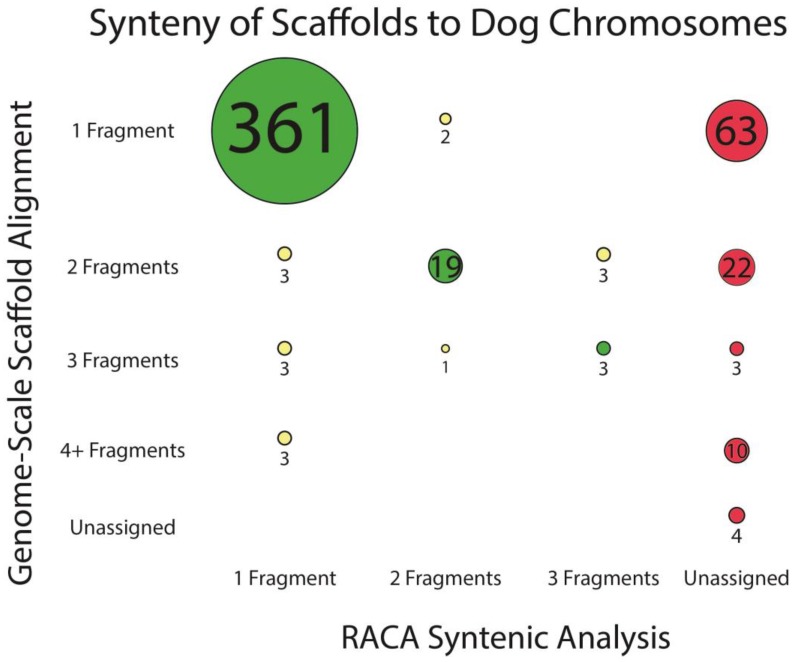
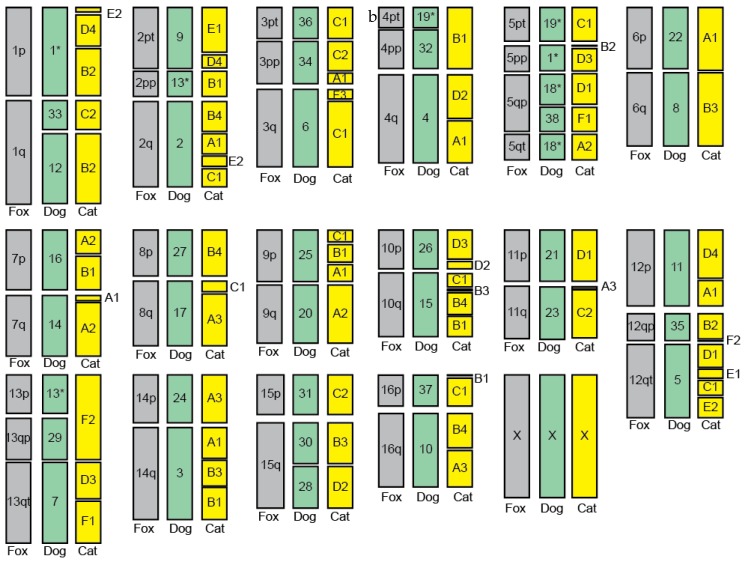
The genome of a red fox () was recently sequenced and assembled using next-generation sequencing (NGS). The assembly is of high quality, with 94X coverage and a scaffold N50 of 11.8 Mbp, but is split into 676,878 scaffolds, some of which are likely to contain assembly errors. Fragmentation and misassembly hinder accurate gene prediction and downstream analysis such as the identification of loci under selection. Therefore, assembly of the genome into chromosome-scale fragments was an important step towards developing this genomic model. Scaffolds from the assembly were aligned to the dog reference genome and compared to the alignment of an outgroup genome (cat) against the dog to identify syntenic sequences among species. The program Reference-Assisted Chromosome Assembly (RACA) then integrated the comparative alignment with the mapping of the raw sequencing reads generated during assembly against the fox scaffolds. The 128 sequence fragments RACA assembled were compared to the fox meiotic linkage map to guide the construction of 40 chromosomal fragments. This computational approach to assembly was facilitated by prior research in comparative mammalian genomics, and the continued improvement of the red fox genome can in turn offer insight into canid and carnivore chromosome evolution. This assembly is also necessary for advancing genetic research in foxes and other canids.
最近利用新一代测序(NGS)技术对赤狐( )的基因组进行了测序和组装。该组装质量很高,覆盖度为94X,支架N50为11.8兆碱基对,但被分成了676,878个支架,其中一些可能包含组装错误。片段化和错误组装阻碍了准确的基因预测以及诸如选择位点识别等下游分析。因此,将基因组组装成染色体规模的片段是开发这个基因组模型的重要一步。组装得到的支架与犬参考基因组进行比对,并与一个外群基因组(猫)与犬的比对结果进行比较,以识别物种间的同线序列。然后,参考辅助染色体组装程序(RACA)将比较比对结果与组装过程中生成的原始测序 reads 与狐狸支架的映射结果整合在一起。将RACA组装的128个序列片段与狐狸减数分裂连锁图谱进行比较,以指导构建40个染色体片段。这种组装的计算方法得益于先前在比较哺乳动物基因组学方面的研究,而赤狐基因组的持续改进反过来又能为犬科动物和食肉动物的染色体进化提供见解。这种组装对于推进狐狸和其他犬科动物的遗传学研究也是必要的。