School of Biomedical and Healthcare Sciences, University of Plymouth, Plymouth, United Kingdom.
School of Sport, Exercise and Rehabilitation Sciences, University of Birmingham, Birmingham, United Kingdom.
PLoS One. 2018 Jun 28;13(6):e0199505. doi: 10.1371/journal.pone.0199505. eCollection 2018.
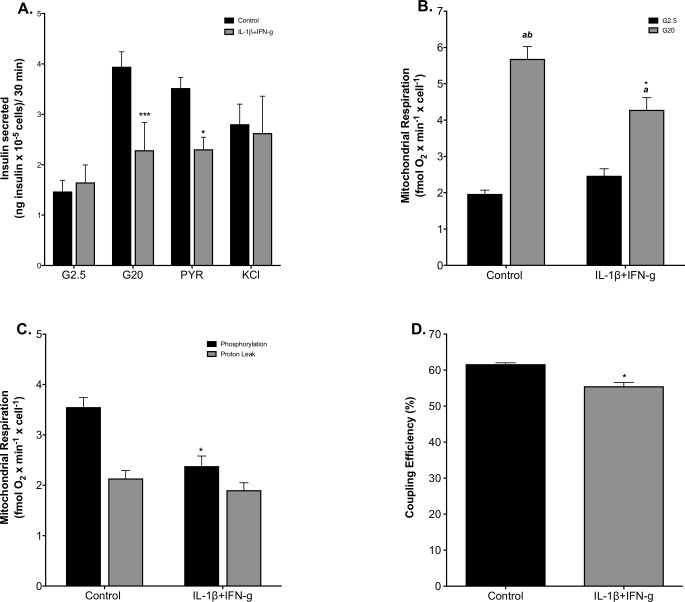
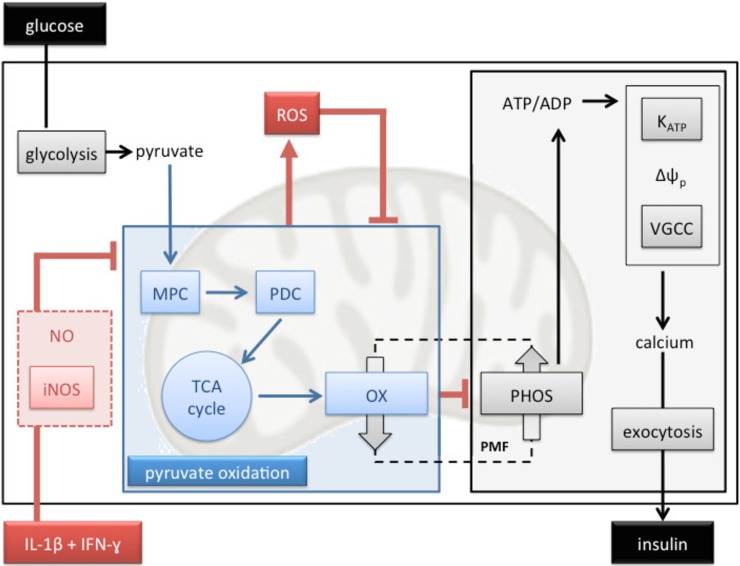
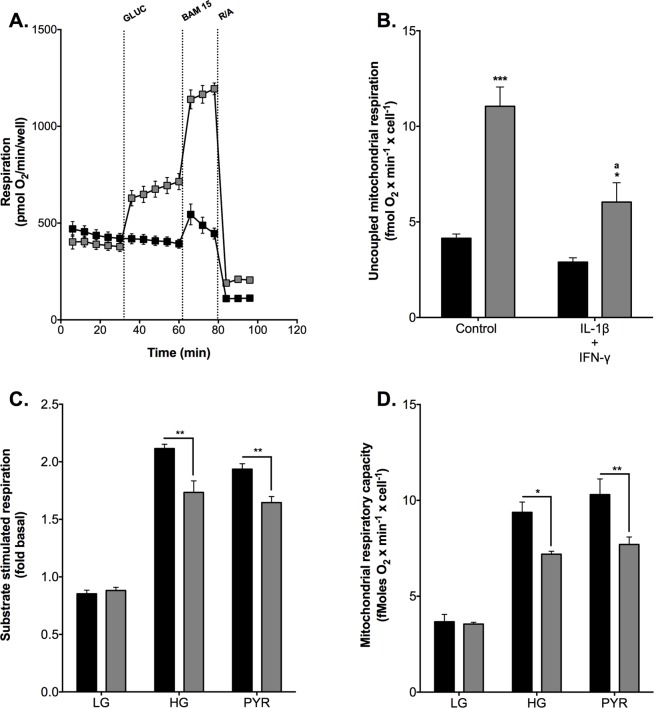
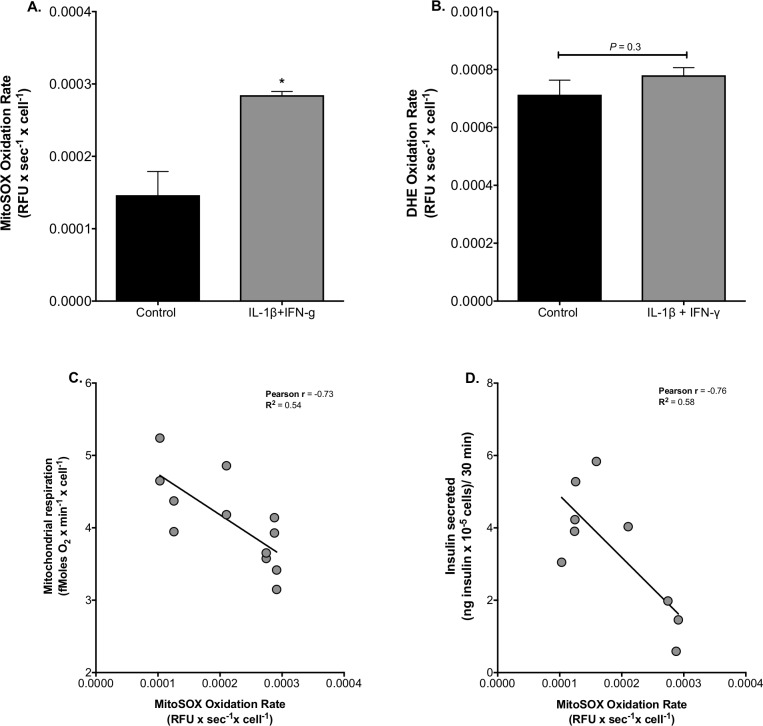
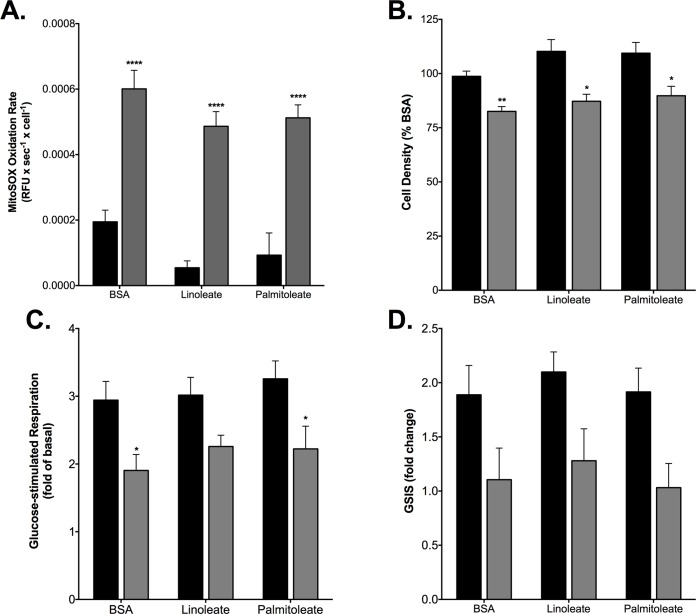
Pro-inflammatory cytokines cause pancreatic beta cell failure during the development of type 2 diabetes. This beta cell failure associates with mitochondrial dysfunction, but the precise effects of cytokines on mitochondrial respiration remain unclear. To test the hypothesis that pro-inflammatory cytokines impair glucose-stimulated insulin secretion (GSIS) by inhibiting oxidative ATP synthesis, we probed insulin release and real-time mitochondrial respiration in rat INS-1E insulinoma cells that were exposed to a combination of 2 ng/mL interleukin-1-beta and 50 ng/mL interferon-gamma. We show that 24-h exposure to these cytokines dampens both glucose- and pyruvate-stimulated insulin secretion (P < 0.0001 and P < 0.05, respectively), but does not affect KCl-induced insulin release. Mirroring secretory defects, glucose- and pyruvate-stimulated mitochondrial respiration are lowered after cytokine exposure (P < 0.01). Further analysis confirms that cytokine-induced mitochondrial respiratory defects occur irrespective of whether fuel oxidation is coupled to, or uncoupled from, ATP synthesis. These observations demonstrate that pro-inflammatory cytokines attenuate GSIS by restricting mitochondrial pyruvate oxidation capacity. Interleukin-1-beta and interferon-gamma also increase mitochondrial superoxide levels (P < 0.05), which may reinforce the inhibition of pyruvate oxidation, and cause a modest (20%) but significant (P < 0.01) loss of INS-1E cells. Cytokine-induced INS-1E cell failure is insensitive to palmitoleate and linoleate, which is at odds with the cytoprotection offered by unsaturated fatty acids against harm caused by nutrient excess. Our data disclose a mitochondrial mechanism for cytokine-impaired GSIS in INS-1E cells, and suggest that inflammatory and nutrient-related beta cell failure emerge, at least partly, through distinct paths.
促炎细胞因子在 2 型糖尿病的发展过程中导致胰岛β细胞衰竭。这种β细胞衰竭与线粒体功能障碍有关,但细胞因子对线粒体呼吸的确切影响尚不清楚。为了验证促炎细胞因子通过抑制氧化型 ATP 合成来损害葡萄糖刺激的胰岛素分泌(GSIS)的假设,我们研究了在暴露于 2ng/ml 白细胞介素-1β和 50ng/ml 干扰素-γ的大鼠 INS-1E 胰岛细胞瘤中胰岛素释放和实时线粒体呼吸。我们发现,这些细胞因子 24 小时暴露会抑制葡萄糖和丙酮酸盐刺激的胰岛素分泌(分别为 P<0.0001 和 P<0.05),但不影响 KCl 诱导的胰岛素释放。与分泌缺陷相似,细胞因子暴露后葡萄糖和丙酮酸盐刺激的线粒体呼吸降低(P<0.01)。进一步分析证实,细胞因子诱导的线粒体呼吸缺陷的发生与燃料氧化是否与 ATP 合成偶联或解偶联无关。这些观察结果表明,促炎细胞因子通过限制线粒体丙酮酸氧化能力来减弱 GSIS。白细胞介素-1β和干扰素-γ还增加线粒体超氧化物水平(P<0.05),这可能加强对丙酮酸氧化的抑制,并导致 INS-1E 细胞发生适度(20%)但显著(P<0.01)的损失。细胞因子诱导的 INS-1E 细胞衰竭对棕榈油酸和亚油酸不敏感,这与不饱和脂肪酸对营养过剩引起的损害提供的细胞保护作用不一致。我们的数据揭示了 INS-1E 细胞中细胞因子诱导的 GSIS 受损的线粒体机制,并表明炎症和与营养相关的β细胞衰竭至少部分通过不同的途径出现。