Institut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona, Maria Aurèlia Capmany i Farnés, 69, 17003 Girona, Catalonia, Spain.
Manchester Institute of Biotechnology, School of Chemical Engineering and Analytical Science, The University of Manchester, 131 Princess Street, Manchester M1 7DN, UK.
Int J Mol Sci. 2018 Jul 6;19(7):1974. doi: 10.3390/ijms19071974.
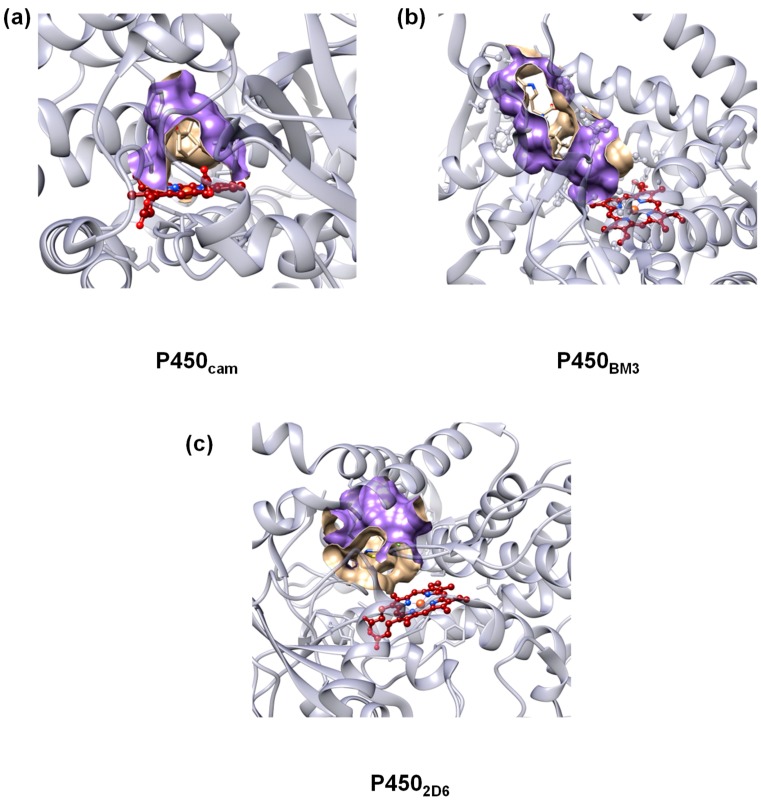
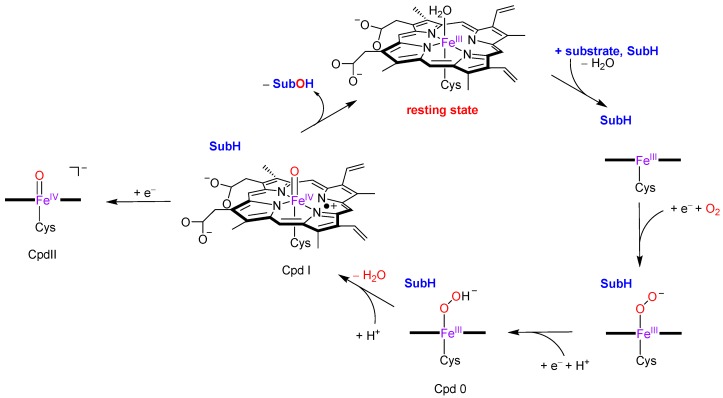
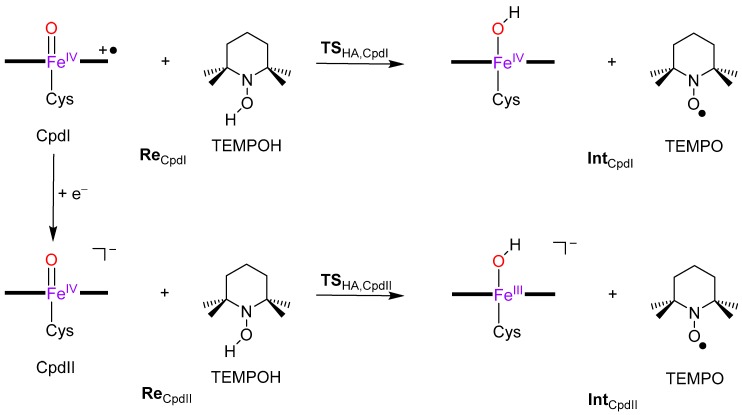
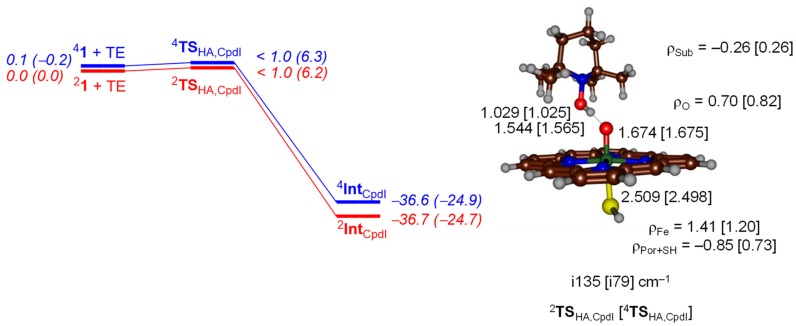
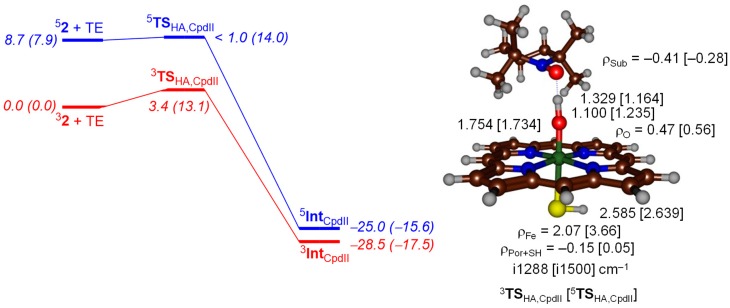
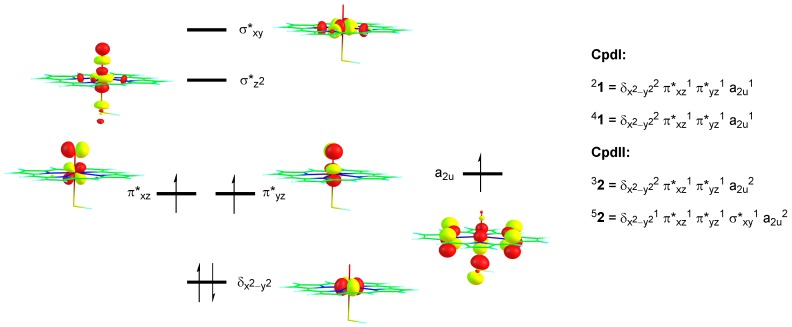
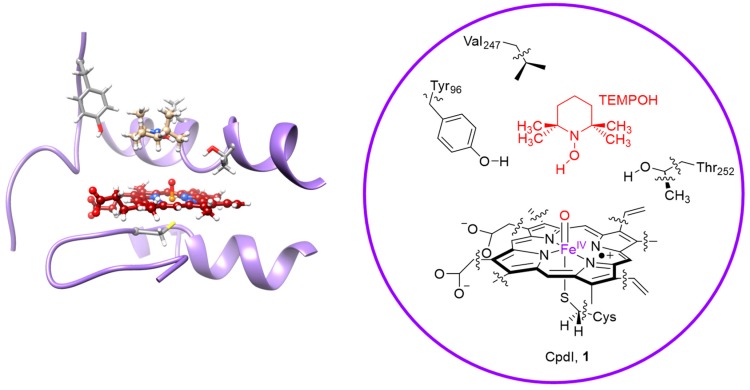
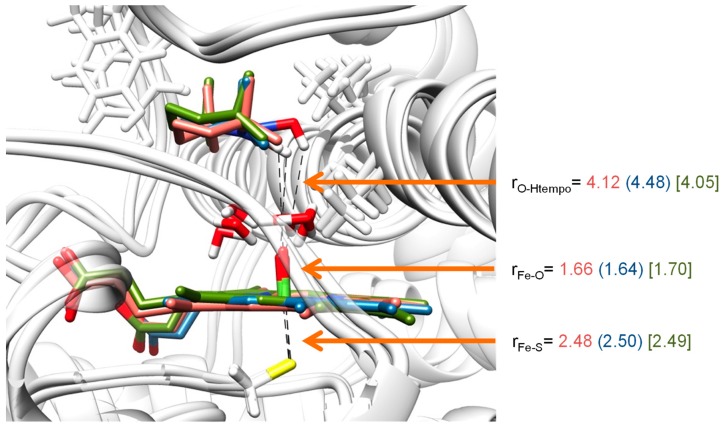
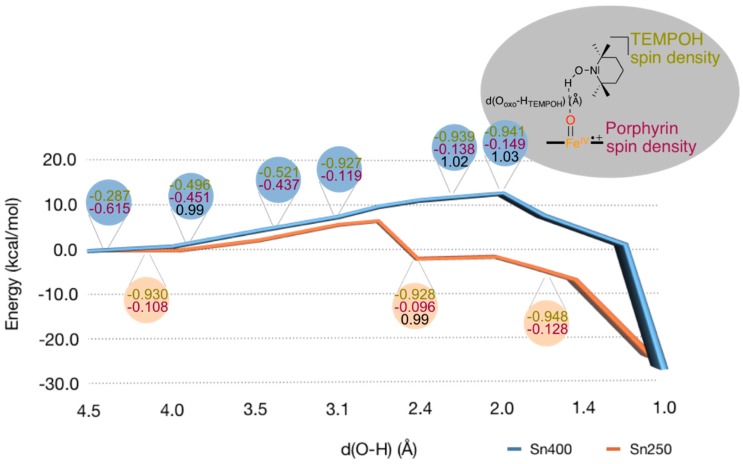
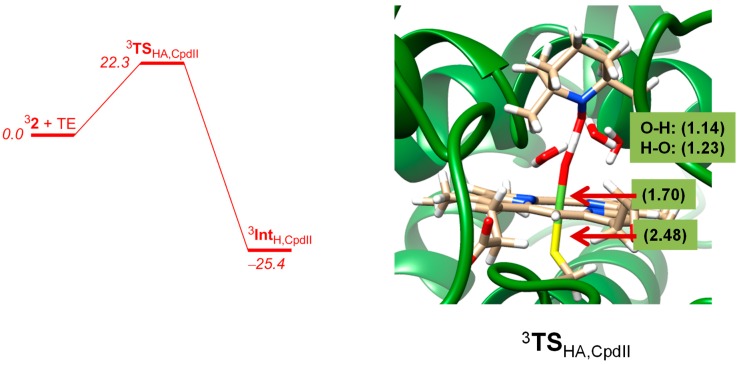
The cytochromes P450 are drug metabolizing enzymes in the body that typically react with substrates through a monoxygenation reaction. During the catalytic cycle two reduction and protonation steps generate a high-valent iron (IV)-oxo heme cation radical species called Compound I. However, with sufficient reduction equivalents present, the catalytic cycle should be able to continue to the reduced species of Compound I, called Compound II, rather than a reaction of Compound I with substrate. In particular, since electron transfer is usually on faster timescales than atom transfer, we considered this process feasible and decided to investigate the reaction computationally. In this work we present a computational study using density functional theory methods on active site model complexes alongside quantum mechanics/molecular mechanics calculations on full enzyme structures of cytochrome P450 enzymes. Specifically, we focus on the relative reactivity of Compound I and II with a model substrate for O⁻H bond activation. We show that generally the barrier heights for hydrogen atom abstraction are higher in energy for Compound II than Compound I for O⁻H bond activation. Nevertheless, for the activation of such bonds, Compound II should still be an active oxidant under enzymatic conditions. As such, our computational modelling predicts that under high-reduction environments the cytochromes P450 can react with substrates via Compound II but the rates will be much slower.
细胞色素 P450 是体内的药物代谢酶,通常通过单加氧反应与底物反应。在催化循环中,两个还原和质子化步骤生成一种称为化合物 I 的高价铁(IV)-氧血红素阳离子自由基物种。然而,当存在足够的还原当量时,催化循环应该能够继续到化合物 I 的还原态,称为化合物 II,而不是化合物 I 与底物的反应。特别是,由于电子转移通常比原子转移快得多,我们认为这个过程是可行的,并决定通过计算来研究这个反应。在这项工作中,我们使用密度泛函理论方法对活性位点模型配合物进行了计算研究,并对细胞色素 P450 酶的全酶结构进行了量子力学/分子力学计算。具体来说,我们专注于化合物 I 和 II 与 O-H 键活化模型底物的相对反应性。我们表明,对于 O-H 键的活化,化合物 II 的氢原子提取的势垒高度通常比化合物 I 高。然而,对于这种键的活化,化合物 II 仍然应该是酶条件下的活性氧化剂。因此,我们的计算模型预测,在高还原环境下,细胞色素 P450 可以通过化合物 II 与底物反应,但速率会慢得多。