State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou, China.
Cell Death Dis. 2018 Jul 9;9(7):753. doi: 10.1038/s41419-018-0794-4.
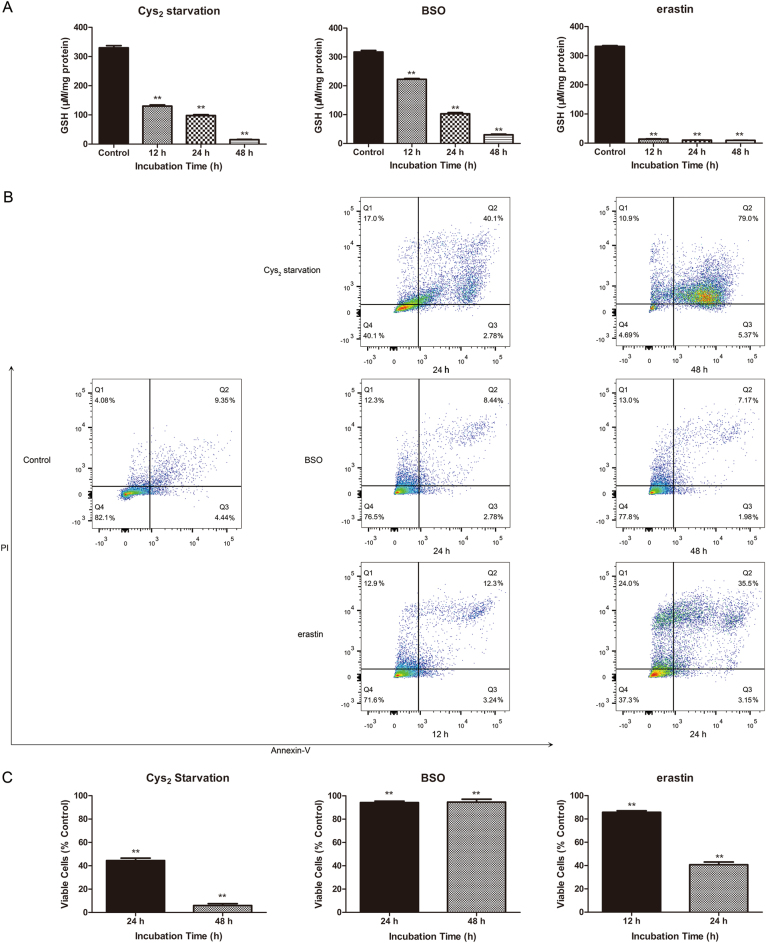
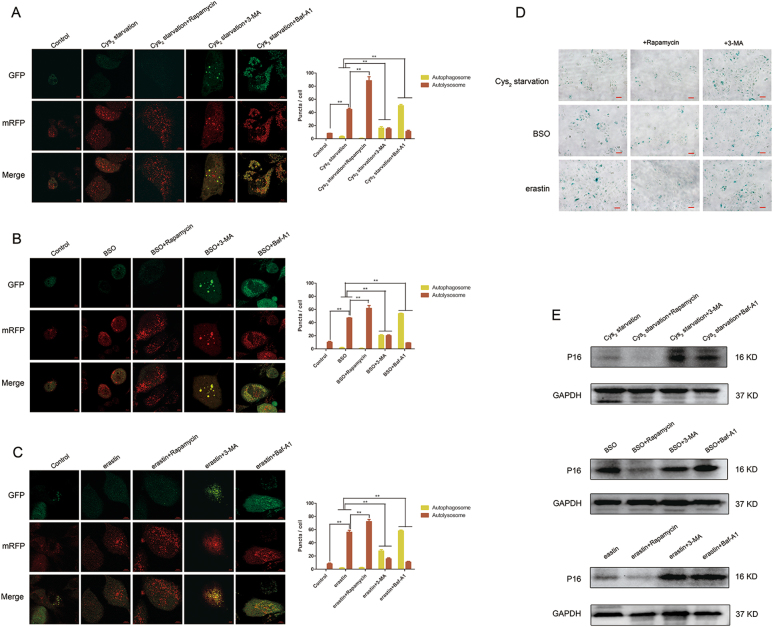
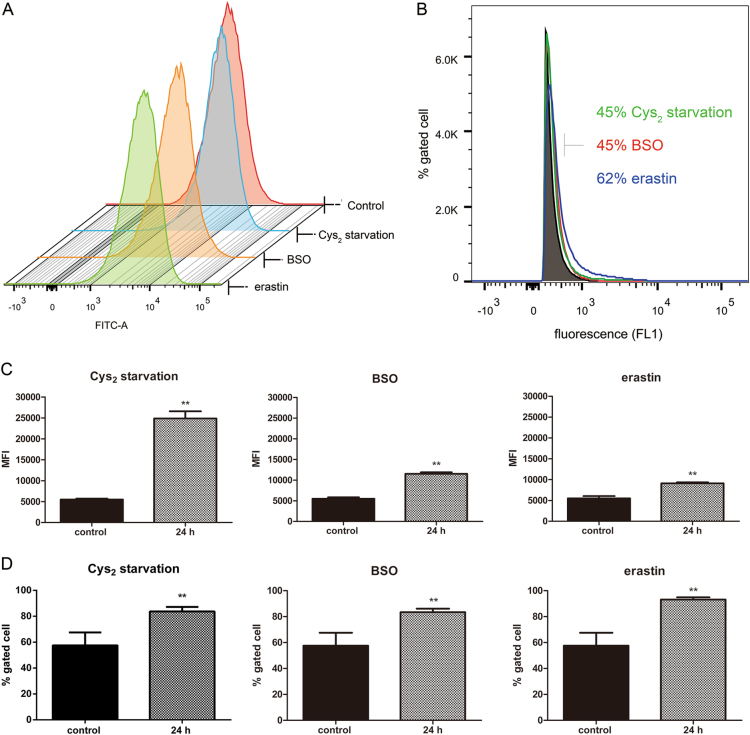
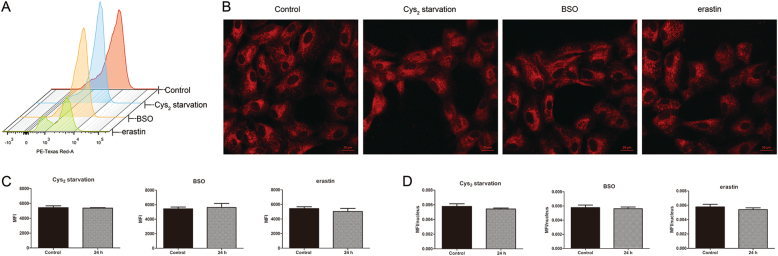
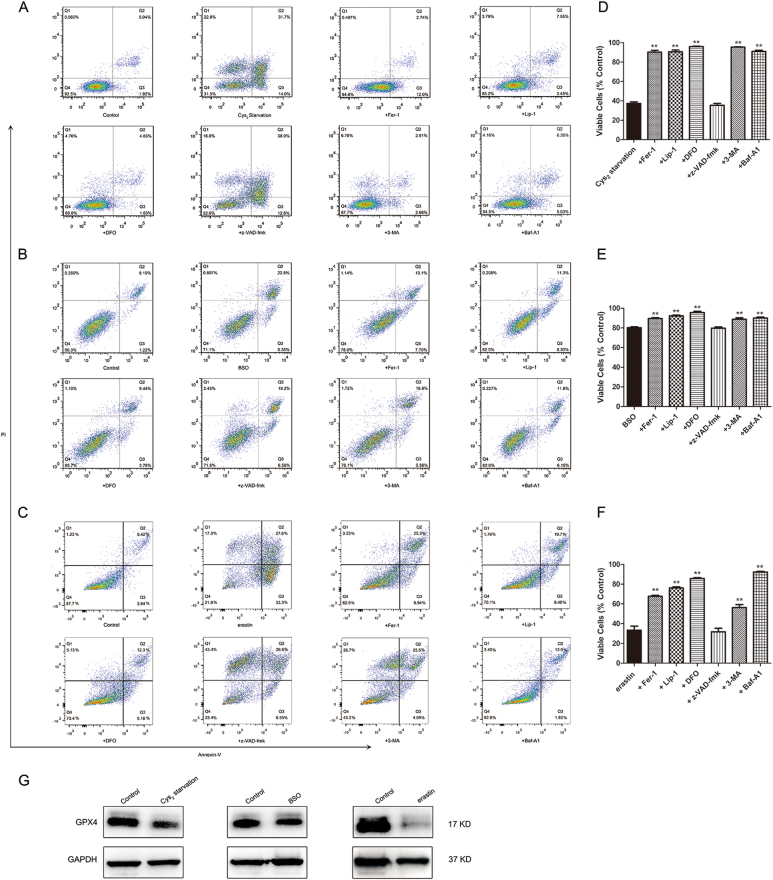
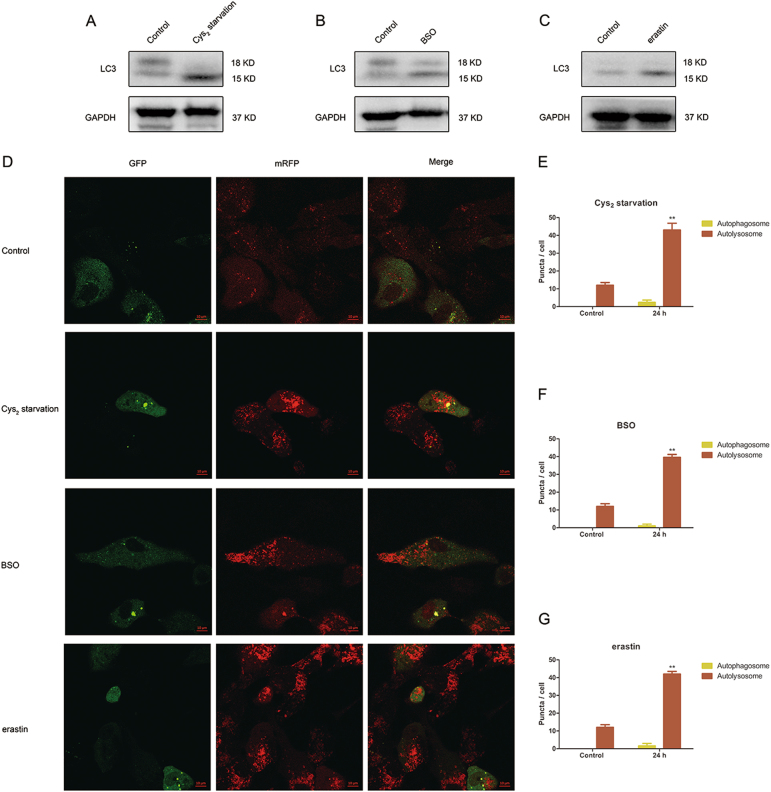
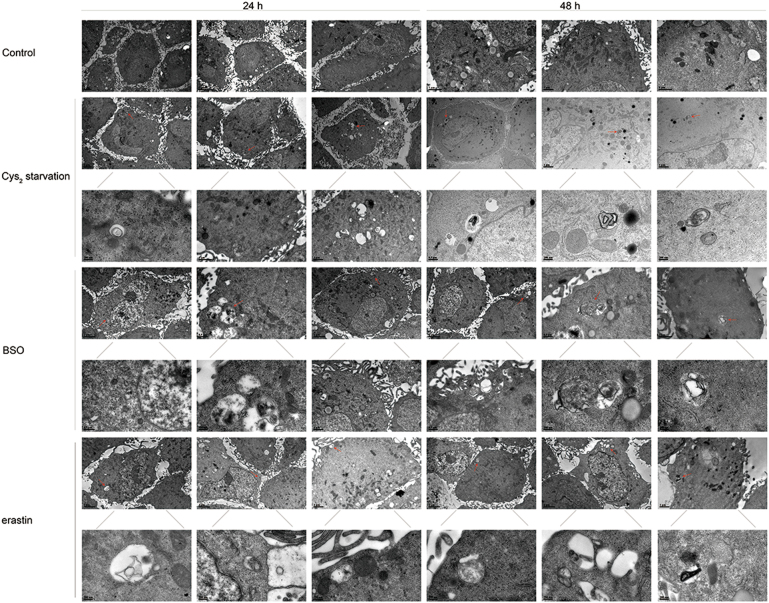
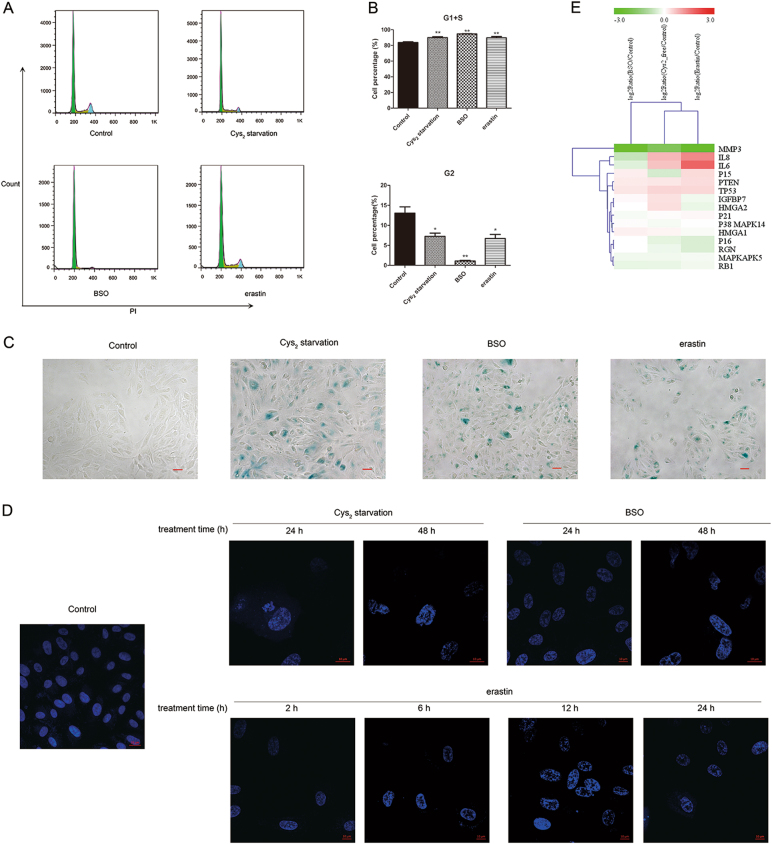
Glutathione (GSH) protects against oxidative damage in many tissues, including retinal pigment epithelium (RPE). Oxidative stress-mediated senescence and death of RPE and subsequent death of photoreceptors have been observed in age-related macular degeneration (AMD). Although the consequences of GSH depletion have been described previously, questions remain regarding the molecular mechanisms. We herein examined the downstream effects of GSH depletion on stress-induced premature senescence (SIPS) and cell death in human RPE cells. Briefly, cultured ARPE-19 cells were depleted of GSH using: (1) incubation in cystine (Cys)-free culture medium; (2) treatment with buthionine sulphoximine (BSO, 1000 µM) to block de novo GSH synthesis for 24-48 h; or (3) treatment with erastin (10 µM for 12-24 h) to inhibit Cys/glutamate antiporter (system x). These treatments decreased cell viability and increased both soluble and lipid reactive oxygen species (ROS) generation but did not affect mitochondrial ROS or mitochondrial mass. Western blot analysis revealed decreased expression of ferroptotic modulator glutathione peroxidase 4 (GPX4). Increased autophagy was apparent, as reflected by increased LC3 expression, autophagic vacuoles, and autophagic flux. In addition, GSH depletion induced SIPS, as evidenced by increased percentage of the senescence-associated β-galactosidase-positive cells, increased senescence-associated heterochromatin foci (SAHF), as well as cell cycle arrest at the G1 phase. GSH depletion-dependent cell death was prevented by selective ferroptosis inhibitors (8 μM Fer-1 and 600 nM Lip-1), iron chelator DFO (80 μM), as well as autophagic inhibitors Baf-A1 (75 nM) and 3-MA (10 mM). Inhibiting autophagy with Baf-A1 (75 nM) or 3-MA (10 mM) promoted SIPS. In contrast, inducing autophagy with rapamycin (100 nM) attenuated SIPS. Our findings suggest that GSH depletion induces ferroptosis, autophagy, and SIPS. In addition, we found that autophagy is activated in the process of ferroptosis and reduces SIPS, suggesting an essential role of autophagy in ferroptosis and SIPS.
谷胱甘肽 (GSH) 可保护许多组织免受氧化损伤,包括视网膜色素上皮 (RPE)。在年龄相关性黄斑变性 (AMD) 中,已观察到氧化应激介导的 RPE 衰老和死亡以及随后的光感受器死亡。尽管以前已经描述了 GSH 耗竭的后果,但关于分子机制仍存在一些问题。在此,我们研究了 GSH 耗竭对人 RPE 细胞应激诱导的过早衰老 (SIPS) 和细胞死亡的下游影响。简要地,用以下方法使培养的 ARPE-19 细胞耗尽 GSH:(1) 在胱氨酸 (Cys)- 缺乏的培养基中孵育;(2) 用丁硫氨酸亚砜胺 (BSO,1000 μM) 处理 24-48 小时以阻断从头 GSH 合成;或 (3) 用 erastin (10 μM 处理 12-24 小时) 抑制半胱氨酸/谷氨酸转运体 (system x)。这些处理降低了细胞活力并增加了可溶性和脂质活性氧物种 (ROS) 的产生,但不影响线粒体 ROS 或线粒体质量。Western blot 分析显示铁死亡调节剂谷胱甘肽过氧化物酶 4 (GPX4) 的表达降低。自噬增加,表现为 LC3 表达增加、自噬小泡和自噬流增加。此外,GSH 耗竭诱导 SIPS,这表现在衰老相关的β-半乳糖苷酶阳性细胞的百分比增加、衰老相关异染色质焦点 (SAHF) 增加以及细胞周期停滞在 G1 期。通过选择性铁死亡抑制剂 (8 μM Fer-1 和 600 nM Lip-1)、铁螯合剂 DFO (80 μM) 以及自噬抑制剂 Baf-A1 (75 nM) 和 3-MA (10 mM) 可预防 GSH 耗竭依赖性细胞死亡。用 Baf-A1 (75 nM) 或 3-MA (10 mM) 抑制自噬会促进 SIPS。相比之下,用雷帕霉素 (100 nM) 诱导自噬会减弱 SIPS。我们的发现表明 GSH 耗竭诱导铁死亡、自噬和 SIPS。此外,我们发现铁死亡过程中激活了自噬,并减少了 SIPS,这表明自噬在铁死亡和 SIPS 中起着重要作用。