1Department of Epidemiology, Johns Hopkins Bloomberg School of Public Health, 615 N. Wolfe Street, Baltimore, MD 21205 USA.
2Wendy Klag Center for Autism and Developmental Disabilities, Johns Hopkins Bloomberg School of Public Health, 615 N. Wolfe Street, W6509, Baltimore, MD 21205 USA.
Mol Autism. 2018 Jun 28;9:40. doi: 10.1186/s13229-018-0224-6. eCollection 2018.
Several reports have suggested a role for epigenetic mechanisms in ASD etiology. Epigenome-wide association studies (EWAS) in autism spectrum disorder (ASD) may shed light on particular biological mechanisms. However, studies of ASD cases versus controls have been limited by post-mortem timing and severely small sample sizes. Reports from in-life sampling of blood or saliva have also been very limited in sample size and/or genomic coverage. We present the largest case-control EWAS for ASD to date, combining data from population-based case-control and case-sibling pair studies.
DNA from 968 blood samples from children in the Study to Explore Early Development (SEED 1) was used to generate epigenome-wide array DNA methylation (DNAm) data at 485,512 CpG sites for 453 cases and 515 controls, using the Illumina 450K Beadchip. The Simons Simplex Collection (SSC) provided 450K array DNAm data on an additional 343 cases and their unaffected siblings. We performed EWAS meta-analysis across results from the two data sets, with adjustment for sex and surrogate variables that reflect major sources of biological variation and technical confounding such as cell type, batch, and ancestry. We compared top EWAS results to those from a previous brain-based analysis. We also tested for enrichment of ASD EWAS CpGs for being targets of meQTL associations using available SNP genotype data in the SEED sample.
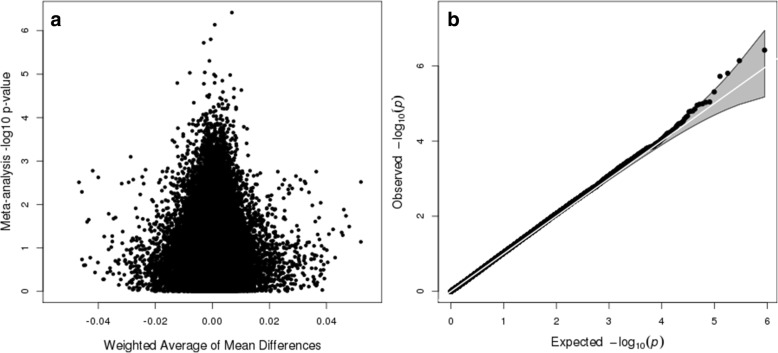
In this meta-analysis of blood-based DNA from 796 cases and 858 controls, no single CpG met a Bonferroni discovery threshold of < 1.12 × 10. Seven CpGs showed differences at < 1 × 10 and 48 at 1 × 10. Of the top 7, 5 showed brain-based ASD associations as well, often with larger effect sizes, and the top 48 overall showed modest concordance ( = 0.31) in direction of effect with cerebellum samples. Finally, we observed suggestive evidence for enrichment of CpG sites controlled by SNPs (meQTL targets) among the EWAS CpG hits, which was consistent across EWAS and meQTL discovery value thresholds.
No single CpG site showed a large enough DNAm difference between cases and controls to achieve epigenome-wide significance in this sample size. However, our results suggest the potential to observe disease associations from blood-based samples. Among the seven sites achieving suggestive statistical significance, we observed consistent, and stronger, effects at the same sites among brain samples. Discovery-oriented EWAS for ASD using blood samples will likely need even larger samples and unified genetic data to further understand DNAm differences in ASD.
多项研究表明,表观遗传机制在自闭症谱系障碍(ASD)的发病机制中起作用。自闭症谱系障碍的全基因组关联研究(EWAS)可能揭示特定的生物学机制。然而,病例对照研究受限于死后时间和严重的小样本量。来自生活中血液或唾液样本的报告在样本量和/或基因组覆盖范围方面也非常有限。我们报告了迄今为止最大的自闭症病例对照全基因组关联研究,该研究结合了基于人群的病例对照和病例-同胞对研究的数据。
来自探索早期发育研究(SEED 1)的 968 名儿童的血液样本中的 DNA 用于生成 453 例病例和 515 例对照的 485512 个 CpG 位点的全基因组表达谱 DNA 甲基化(DNAm)数据,使用的是 Illumina 450K Beadchip。西蒙斯单倍型收集(SSC)提供了 343 例病例及其未受影响的兄弟姐妹的 450K 阵列 DNAm 数据。我们对来自两个数据集的结果进行了 EWAS 元分析,并进行了性别和替代变量的调整,这些替代变量反映了主要的生物学变异和技术混杂来源,如细胞类型、批次和祖源。我们将顶级 EWAS 结果与之前基于大脑的分析进行了比较。我们还测试了可用的 SEED 样本中 SNP 基因型数据是否富集了 ASD EWAS CpGs 作为 meQTL 关联的靶点。
在这项基于血液的 796 例病例和 858 例对照的 DNA 的荟萃分析中,没有一个 CpG 达到了 Bonferroni 发现阈值<1.12×10。有 7 个 CpG 在<1×10时有差异,48 个在 1×10时有差异。在顶部 7 个中,5 个在大脑中也显示出了自闭症的关联,通常具有更大的效应大小,而前 48 个总体上与小脑样本的效应方向具有适度的一致性(=0.31)。最后,我们观察到在 EWAS CpG 命中存在提示性证据,表明 CpG 位点受 SNP(meQTL 靶点)控制的富集,这在 EWAS 和 meQTL 发现值阈值上是一致的。
在这个样本量中,没有一个 CpG 位点的 DNAm 差异大到足以达到全基因组关联研究的显著性水平。然而,我们的结果表明,从基于血液的样本中观察到疾病关联的可能性。在达到提示性统计显著性的七个位点中,我们在大脑样本中观察到了相同位点的一致且更强的效应。使用血液样本进行以发现为导向的自闭症全基因组关联研究可能需要更大的样本量和统一的遗传数据,以进一步了解自闭症中的 DNAm 差异。