Department of Radiation Physics, Institute of Clinical Sciences, Sahlgrenska Cancer Center, Sahlgrenska Academy at the University of Gothenburg, Sahlgrenska University Hospital, Gothenburg, Sweden.
Department of Applied Physics, Chalmers University of Technology, Gothenburg, Sweden.
PLoS One. 2018 Jul 12;13(7):e0197911. doi: 10.1371/journal.pone.0197911. eCollection 2018.
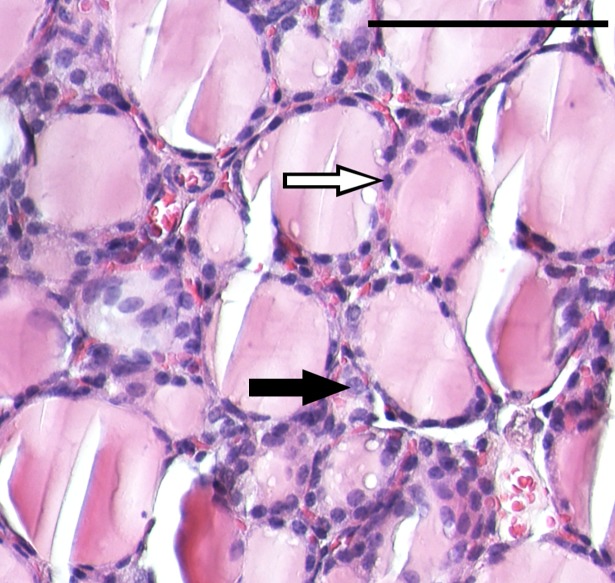
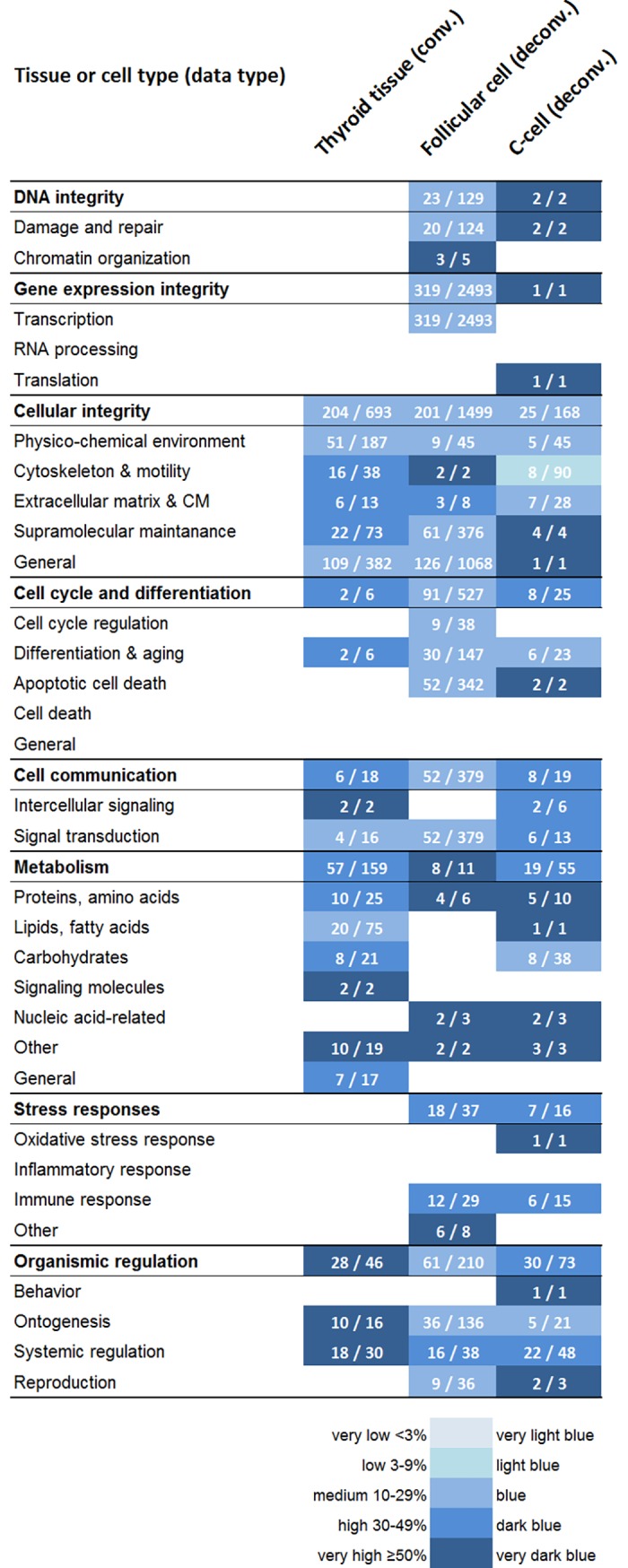
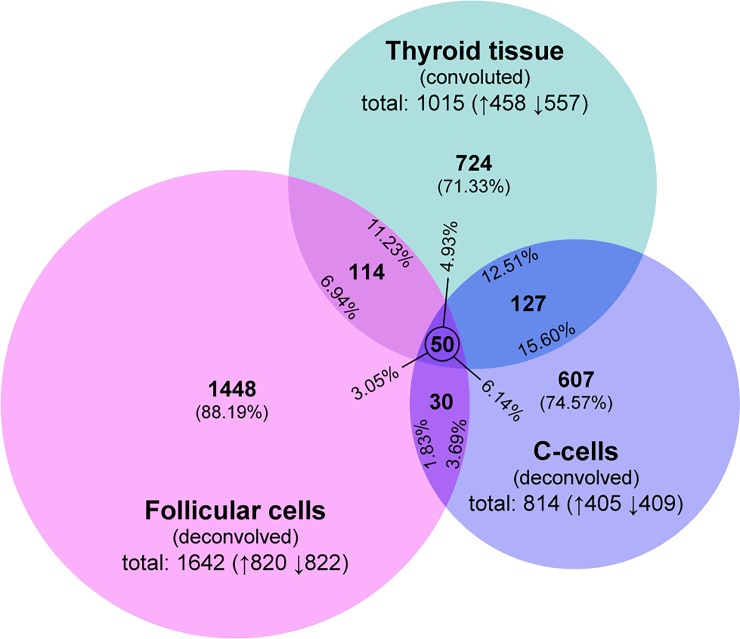
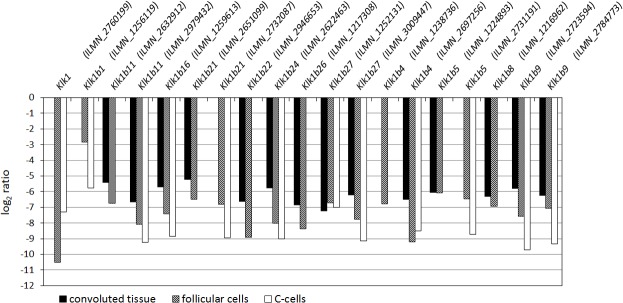
High-throughput gene expression analysis is increasingly used in radiation research for discovery of damage-related or absorbed dose-dependent biomarkers. In tissue samples, cell type-specific responses can be masked in expression data due to mixed cell populations which can preclude biomarker discovery. In this study, we deconvolved microarray data from thyroid tissue in order to assess possible bias from mixed cell type data. Transcript expression data [GSE66303] from mouse thyroid that received 5.9 Gy from 131I over 24 h (or 0 Gy from mock treatment) were deconvolved by cell frequency of follicular cells and C-cells using csSAM and R and processed with Nexus Expression. Literature-based signature genes were used to assess the relative impact from ionizing radiation (IR) or thyroid hormones (TH). Regulation of cellular functions was inferred by enriched biological processes according to Gene Ontology terms. We found that deconvolution increased the detection rate of significantly regulated transcripts including the biomarker candidate family of kallikrein transcripts. Detection of IR-associated and TH-responding signature genes was also increased in deconvolved data, while the dominating trend of TH-responding genes was reproduced. Importantly, responses in biological processes for DNA integrity, gene expression integrity, and cellular stress were not detected in convoluted data-which was in disagreement with expected dose-response relationships-but upon deconvolution in follicular cells and C-cells. In conclusion, previously reported trends of 131I-induced transcriptional responses in thyroid were reproduced with deconvolved data and usually with a higher detection rate. Deconvolution also resolved an issue with detecting damage and stress responses in enriched data, and may reduce false negatives in other contexts as well. These findings indicate that deconvolution can optimize microarray data analysis of heterogeneous sample material for biomarker screening or other clinical applications.
高通量基因表达分析越来越多地应用于辐射研究中,以发现与损伤相关或与吸收剂量相关的生物标志物。在组织样本中,由于混合细胞群体,细胞类型特异性反应可能在表达数据中被掩盖,从而阻碍生物标志物的发现。在这项研究中,我们对甲状腺组织的微阵列数据进行了去卷积处理,以评估混合细胞类型数据可能带来的偏差。使用 csSAM 和 R 对来自接受 131I 24 小时内 5.9 Gy(或模拟处理 0 Gy)辐射的小鼠甲状腺的转录表达数据[GSE66303]进行了基于细胞频率的滤泡细胞和 C 细胞去卷积处理,并使用 Nexus Expression 进行了处理。使用基于文献的特征基因来评估电离辐射(IR)或甲状腺激素(TH)的相对影响。根据基因本体论术语,通过丰富的生物学过程来推断细胞功能的调节。我们发现,去卷积增加了显著调节转录物的检测率,包括 kallikrein 转录物的生物标志物候选家族。去卷积数据中也增加了 IR 相关和 TH 响应的特征基因的检测,而 TH 响应基因的主导趋势得到了重现。重要的是,在卷积数据中未检测到与 DNA 完整性、基因表达完整性和细胞应激相关的生物学过程反应,这与预期的剂量反应关系不一致,但在滤泡细胞和 C 细胞中去卷积后则可以检测到。总之,使用去卷积数据重现了 131I 诱导的甲状腺转录反应的先前报道趋势,并且通常具有更高的检测率。去卷积还解决了在富集数据中检测损伤和应激反应的问题,并且在其他情况下也可能减少假阴性。这些发现表明,去卷积可以优化异质样本材料的微阵列数据分析,用于生物标志物筛选或其他临床应用。