Institute for Molecular Biosciences, University of Queensland, Brisbane, Australia.
BMC Bioinformatics. 2018 Jul 16;19(1):267. doi: 10.1186/s12859-018-2282-3.
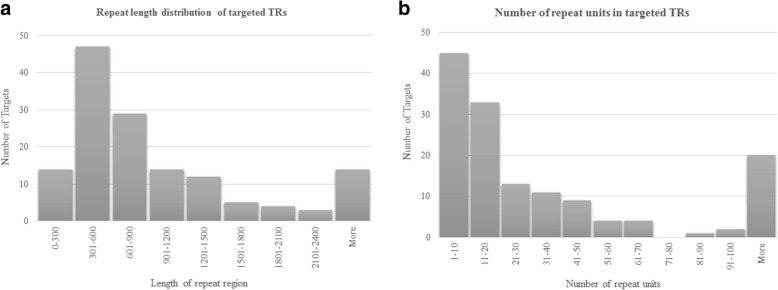
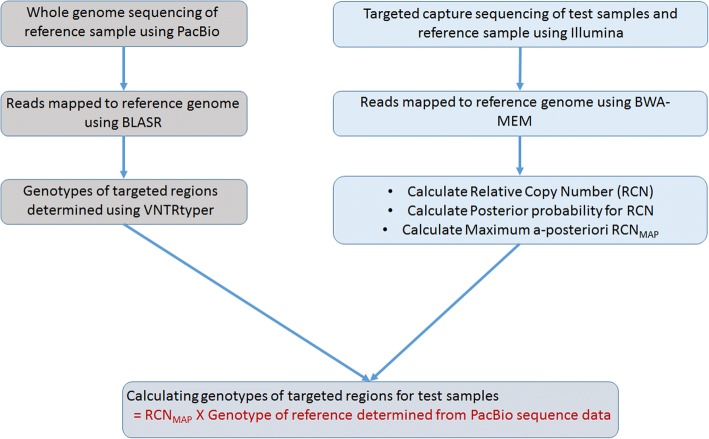
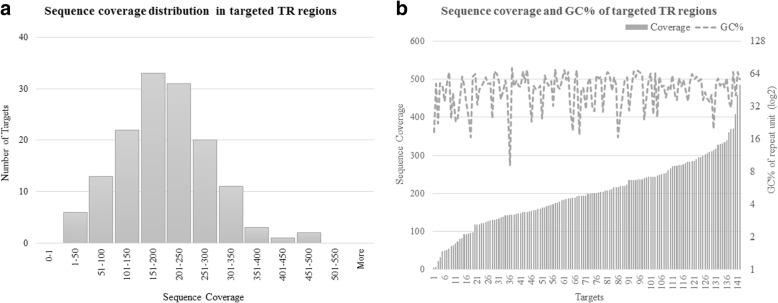
Tandem repeats comprise significant proportion of the human genome including coding and regulatory regions. They are highly prone to repeat number variation and nucleotide mutation due to their repetitive and unstable nature, making them a major source of genomic variation between individuals. Despite recent advances in high throughput sequencing, analysis of tandem repeats in the context of complex diseases is still hindered by technical limitations. We report a novel targeted sequencing approach, which allows simultaneous analysis of hundreds of repeats. We developed a Bayesian algorithm, namely - GtTR - which combines information from a reference long-read dataset with a short read counting approach to genotype tandem repeats at population scale. PCR sizing analysis was used for validation.
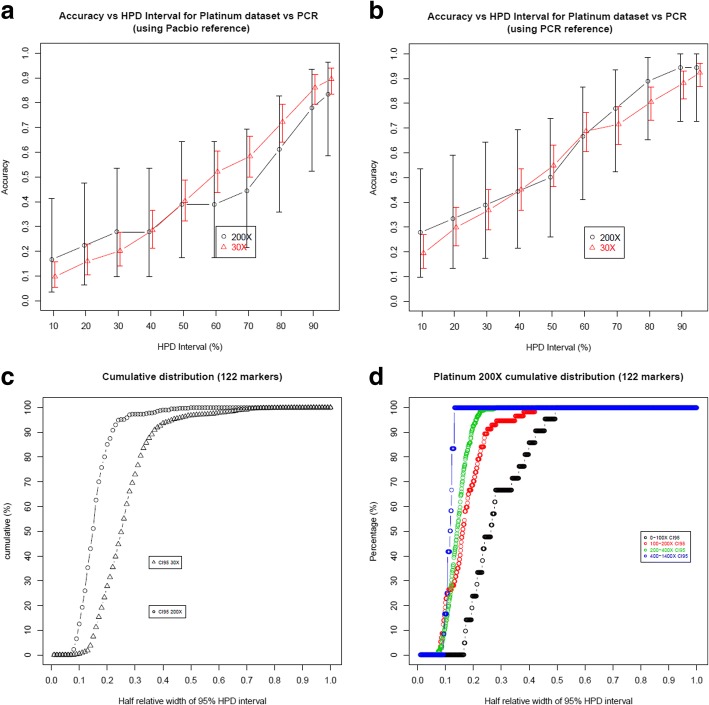
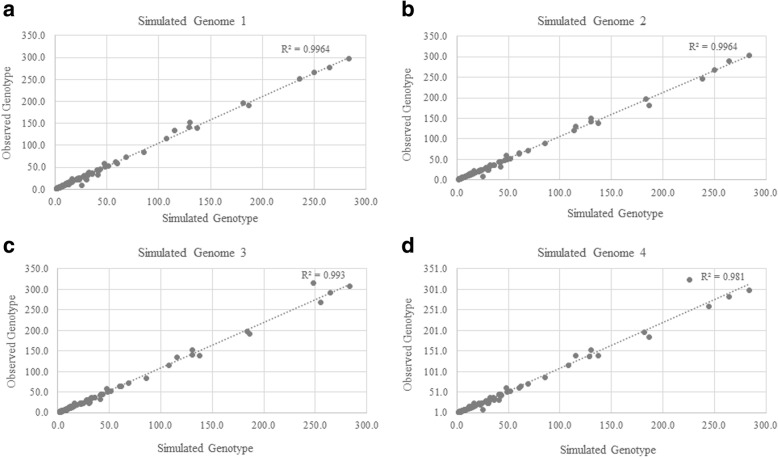
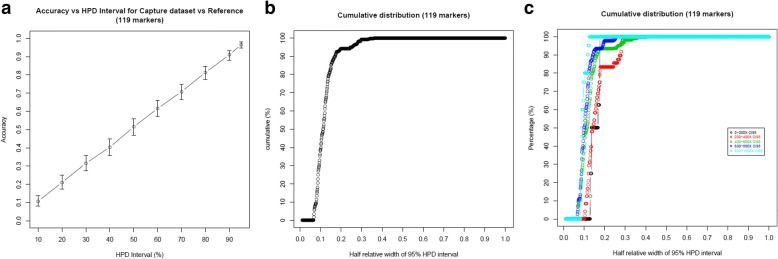
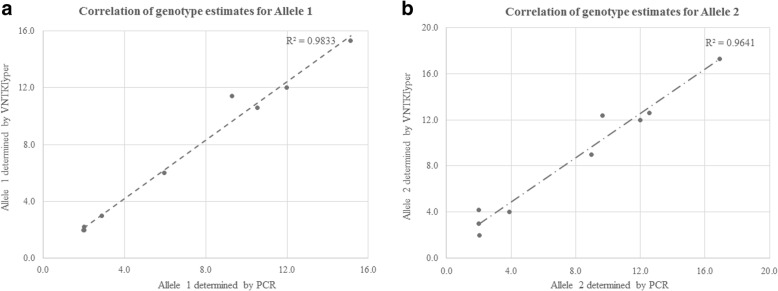
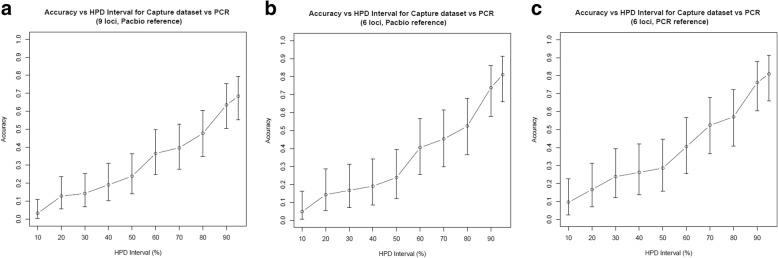
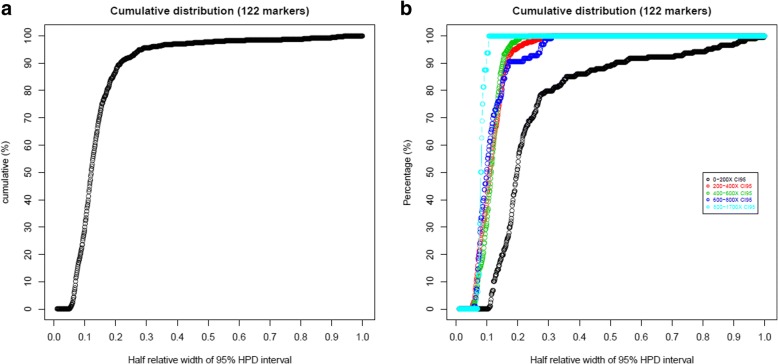
We used a PacBio long-read sequenced sample to generate a reference tandem repeat genotype dataset with on average 13% absolute deviation from PCR sizing results. Using this reference dataset GtTR generated estimates of VNTR copy number with accuracy within 95% high posterior density (HPD) intervals of 68 and 83% for capture sequence data and 200X WGS data respectively, improving to 87 and 94% with use of a PCR reference. We show that the genotype resolution increases as a function of depth, such that the median 95% HPD interval lies within 25, 14, 12 and 8% of the its midpoint copy number value for 30X, 200X WGS, 395X and 800X capture sequence data respectively. We validated nine targets by PCR sizing analysis and genotype estimates from sequencing results correlated well with PCR results.
The novel genotyping approach described here presents a new cost-effective method to explore previously unrecognized class of repeat variation in GWAS studies of complex diseases at the population level. Further improvements in accuracy can be obtained by improving accuracy of the reference dataset.
串联重复序列构成了人类基因组的重要部分,包括编码区和调控区。由于其重复和不稳定的性质,它们非常容易发生重复数量的变化和核苷酸突变,这使得它们成为个体之间基因组变异的主要来源。尽管高通量测序技术取得了最近的进展,但在复杂疾病背景下分析串联重复序列仍然受到技术限制的阻碍。我们报告了一种新的靶向测序方法,该方法允许同时分析数百个重复序列。我们开发了一种贝叶斯算法,即 GtTR,它结合了来自参考长读数据集的信息和短读计数方法,以在群体水平上对串联重复序列进行基因分型。PCR 大小分析用于验证。
我们使用 PacBio 长读测序样本生成了一个参考串联重复基因型数据集,其平均绝对偏差为 PCR 大小分析结果的 13%。使用该参考数据集,GtTR 生成的 VNTR 拷贝数估计值在捕获序列数据和 200X WGS 数据中的准确度分别在 68%和 83%的 95%高后验密度(HPD)区间内,使用 PCR 参考值可提高到 87%和 94%。我们表明,基因型分辨率随深度增加而增加,使得中位 95%HPD 区间在 30X、200X WGS、395X 和 800X 捕获序列数据中分别为其中点拷贝数值的 25%、14%、12%和 8%。我们通过 PCR 大小分析验证了九个靶标,测序结果的基因型估计与 PCR 结果相关性良好。
这里描述的新型基因分型方法提供了一种新的具有成本效益的方法,可在复杂疾病的 GWAS 研究中探索以前未被识别的重复变异类别,达到群体水平。通过提高参考数据集的准确性,可以进一步提高准确性。