Illumina Inc., San Diego, CA, 92122, USA.
Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, USA.
Genome Med. 2022 Aug 11;14(1):84. doi: 10.1186/s13073-022-01085-z.
Expansions of short tandem repeats are the cause of many neurogenetic disorders including familial amyotrophic lateral sclerosis, Huntington disease, and many others. Multiple methods have been recently developed that can identify repeat expansions in whole genome or exome sequencing data. Despite the widely recognized need for visual assessment of variant calls in clinical settings, current computational tools lack the ability to produce such visualizations for repeat expansions. Expanded repeats are difficult to visualize because they correspond to large insertions relative to the reference genome and involve many misaligning and ambiguously aligning reads.
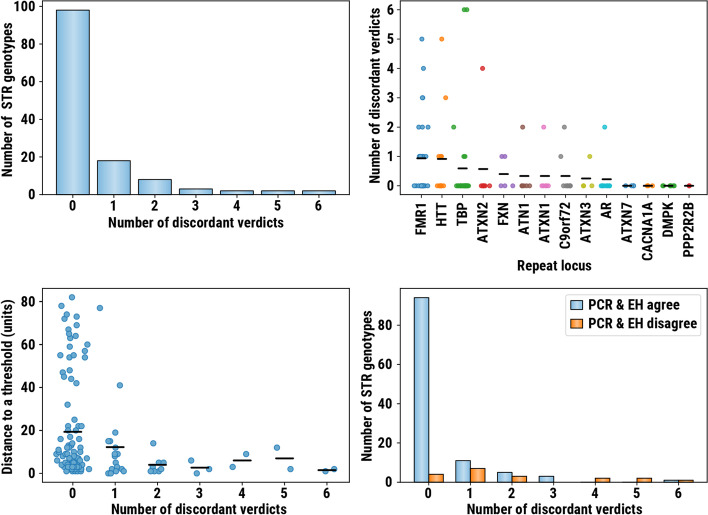
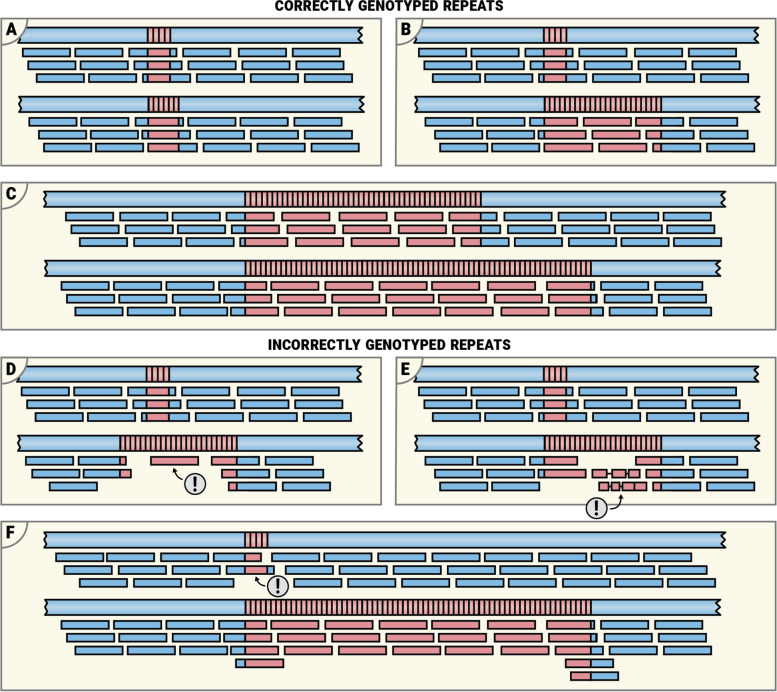
We implemented REViewer, a computational method for visualization of sequencing data in genomic regions containing long repeat expansions and FlipBook, a companion image viewer designed for manual curation of large collections of REViewer images. To generate a read pileup, REViewer reconstructs local haplotype sequences and distributes reads to these haplotypes in a way that is most consistent with the fragment lengths and evenness of read coverage. To create appropriate training materials for onboarding new users, we performed a concordance study involving 12 scientists involved in short tandem repeat research. We used the results of this study to create a user guide that describes the basic principles of using REViewer as well as a guide to the typical features of read pileups that correspond to low confidence repeat genotype calls. Additionally, we demonstrated that REViewer can be used to annotate clinically relevant repeat interruptions by comparing visual assessment results of 44 FMR1 repeat alleles with the results of triplet repeat primed PCR. For 38 of these alleles, the results of visual assessment were consistent with triplet repeat primed PCR.
Read pileup plots generated by REViewer offer an intuitive way to visualize sequencing data in regions containing long repeat expansions. Laboratories can use REViewer and FlipBook to assess the quality of repeat genotype calls as well as to visually detect interruptions or other imperfections in the repeat sequence and the surrounding flanking regions. REViewer and FlipBook are available under open-source licenses at https://github.com/illumina/REViewer and https://github.com/broadinstitute/flipbook respectively.
短串联重复序列的扩增是许多神经遗传疾病的病因,包括家族性肌萎缩侧索硬化症、亨廷顿病和许多其他疾病。最近开发了多种方法,可以在全基因组或外显子组测序数据中识别重复扩增。尽管在临床环境中广泛认识到需要对变异呼叫进行视觉评估,但当前的计算工具缺乏为重复扩展生成此类可视化效果的能力。扩展重复难以可视化,因为它们相对于参考基因组来说是较大的插入,并且涉及许多错配和模糊对齐的读取。
我们实现了 REViewer,这是一种用于可视化基因组区域中包含长重复扩展的测序数据的计算方法,以及 FlipBook,这是一种专为手动管理大量 REViewer 图像而设计的图像查看器。为了生成读取堆积,REViewer 重建局部单倍型序列,并以最符合片段长度和读取覆盖均匀性的方式将读取分配给这些单倍型。为了为新用户提供入职培训材料,我们进行了一项涉及 12 名从事短串联重复研究的科学家的一致性研究。我们使用这项研究的结果创建了一个用户指南,描述了使用 REViewer 的基本原理,以及与低置信度重复基因型呼叫相对应的读取堆积的典型特征的指南。此外,我们通过将 44 个 FMR1 重复等位基因的视觉评估结果与三核苷酸重复引物 PCR 的结果进行比较,证明了 REViewer 可用于注释临床相关的重复中断。在这些等位基因中,有 38 个的结果与三核苷酸重复引物 PCR 一致。
由 REViewer 生成的读取堆积图提供了一种直观的方法,可以可视化包含长重复扩展的区域中的测序数据。实验室可以使用 REViewer 和 FlipBook 来评估重复基因型呼叫的质量,以及通过视觉检测重复序列和周围侧翼区域中的中断或其他缺陷。REViewer 和 FlipBook 可在以下网址以开源许可证获得:https://github.com/illumina/REViewer 和 https://github.com/broadinstitute/flipbook。