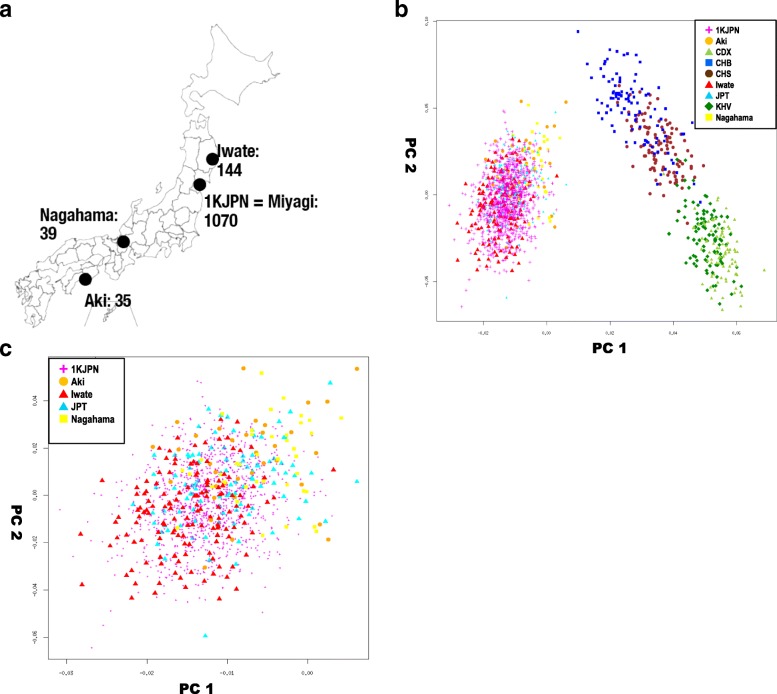
Sendai, Tohoku Medical Megabank Organization, 2-1, Seiryo-machi, Aoba-ku, Tohoku Medical Megabank, Tohoku University, Sendai, 980-8573, Miyagi, Japan.
Present address: Department of Human Genetics, Graduate School of Medicine, The University of Tokyo, Bunkyo-ku, 113-0033, Tokyo, Japan.
BMC Genomics. 2018 Jul 24;19(1):551. doi: 10.1186/s12864-018-4942-0.
Genotype imputation from single-nucleotide polymorphism (SNP) genotype data using a haplotype reference panel consisting of thousands of unrelated individuals from populations of interest can help to identify strongly associated variants in genome-wide association studies. The Tohoku Medical Megabank (TMM) project was established to support the development of precision medicine, together with the whole-genome sequencing of 1070 human genomes from individuals in the Miyagi region (Northeast Japan) and the construction of the 1070 Japanese genome reference panel (1KJPN). Here, we investigated the performance of 1KJPN for genotype imputation of Japanese samples not included in the TMM project and compared it with other population reference panels.
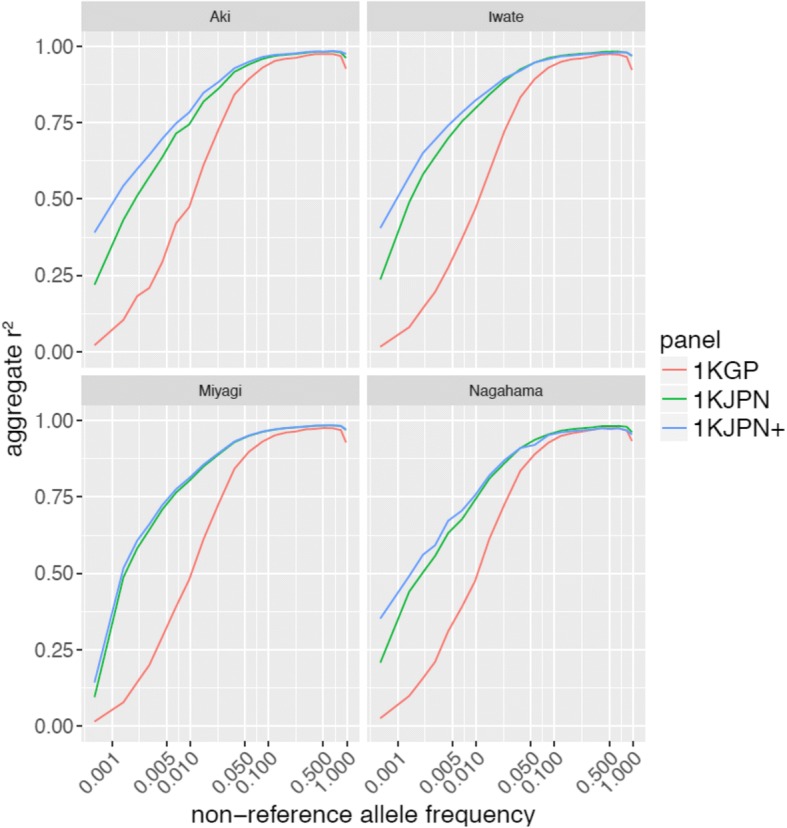
We found that the 1KJPN population was more similar to other Japanese populations, Nagahama (south-central Japan) and Aki (Shikoku Island), than to East Asian populations in the 1000 Genomes Project other than JPT, suggesting that the large-scale collection (more than 1000) of Japanese genomes from the Miyagi region covered many of the genetic variations of Japanese in mainland Japan. Moreover, 1KJPN outperformed the phase 3 reference panel of the 1000 Genomes Project (1KGPp3) for Japanese samples, and IKJPN showed similar imputation rates for the TMM and other Japanese samples for SNPs with minor allele frequencies (MAFs) higher than 1%.
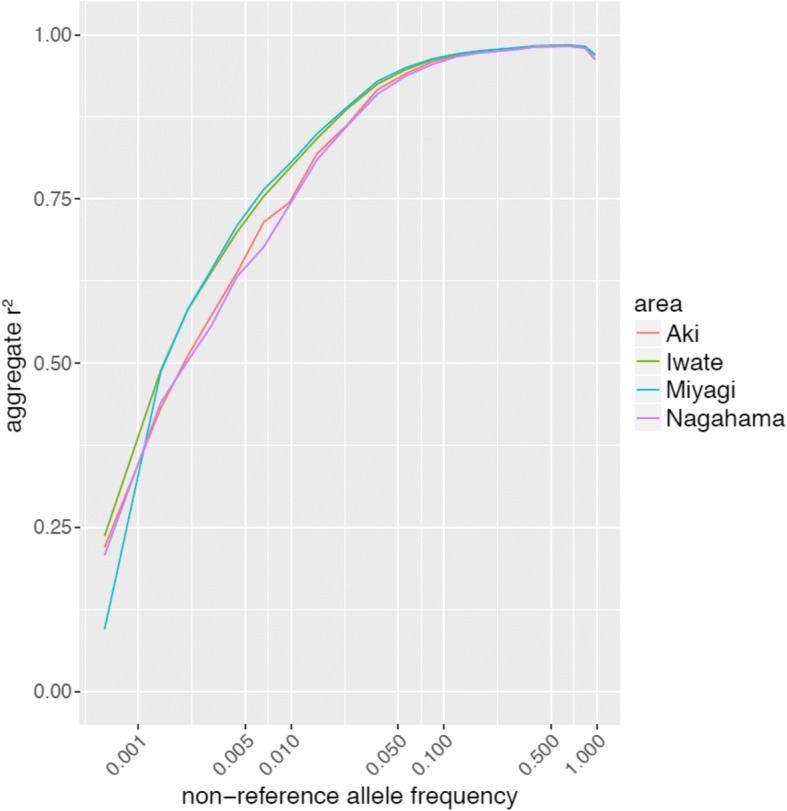
1KJPN covered most of the variants found in the samples from areas of the Japanese mainland outside the Miyagi region, implying 1KJPN is representative of the Japanese population's genomes. 1KJPN and successive reference panels are useful genome reference panels for the mainland Japanese population. Importantly, the addition of whole genome sequences not included in the 1KJPN panel improved imputation efficiencies for SNPs with MAFs under 1% for samples from most regions of the Japanese archipelago.
使用包含来自目标人群中数千个无关个体的单核苷酸多态性 (SNP) 基因型数据的单体型参考面板,对单核苷酸多态性基因型数据进行基因分型,可以帮助在全基因组关联研究中识别与疾病关联较强的变体。东北医疗大数据项目 (Tohoku Medical Megabank,TMM) 与 1070 个人类基因组的全基因组测序项目同时建立,这些人类基因组来自日本宫城县(日本东北部)的个体,同时构建了 1070 个日本人基因组参考面板(1KJPN)。在这里,我们研究了 1KJPN 对 TMM 项目以外的日本样本的基因分型性能,并将其与其他人群参考面板进行了比较。
我们发现 1KJPN 人群与日本其他人群,包括日本中部的长滨(Nagahama)和四国岛的安芸(Aki),比除 JPT 以外的 1000 基因组计划中的东亚人群更为相似,这表明从宫城县大规模收集(超过 1000 个)的日本基因组涵盖了日本本土日本人群的许多遗传变异。此外,1KJPN 对日本样本的 1000 基因组计划第 3 阶段参考面板(1KGPp3)的表现优于后者,对于 MAF 高于 1%的 SNP,1KJPN 对 TMM 和其他日本样本的基因分型率相似。
1KJPN 涵盖了宫城县以外的日本本土地区样本中发现的大多数变体,这意味着 1KJPN 代表了日本人群的基因组。1KJPN 和后续的参考面板是日本大陆人群有用的基因组参考面板。重要的是,添加不属于 1KJPN 面板的全基因组序列可以提高 MAF 低于 1%的 SNP 在大多数日本群岛地区样本中的基因分型效率。