Department of Paediatric Growth, Diabetes and Endocrinology, National Children's Hospital, Tallaght University Hospital, Dublin, Ireland.
Trinity College Dublin, The University of Dublin, Dublin, Ireland.
Clin Endocrinol (Oxf). 2018 Dec;89(6):813-823. doi: 10.1111/cen.13827. Epub 2018 Oct 1.
Loss-of-function mutations in IGSF1 result in X-linked central congenital hypothyroidism (CeCH), occurring in isolation or associated with additional pituitary hormone deficits. Intrafamilial penetrance is highly variable and a minority of heterozygous females are also affected. We identified and characterized a novel IGSF1 mutation and investigated its associated phenotypes in a large Irish kindred.
DESIGN, PATIENTS AND MEASUREMENTS: A novel hemizygous IGSF1 mutation was identified by direct sequencing in two brothers with CeCH, and its functional consequences were characterized in vitro. Genotype-phenotype correlations were investigated in the wider kindred.
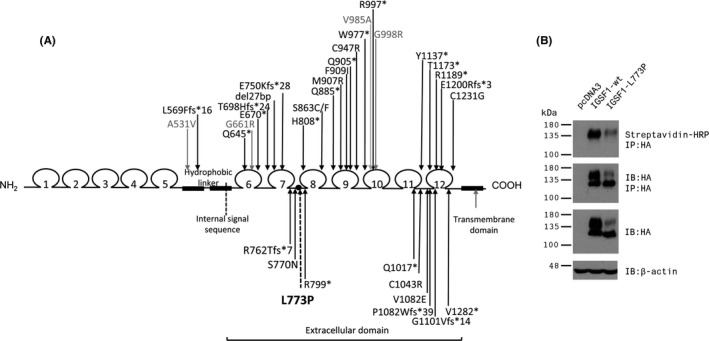
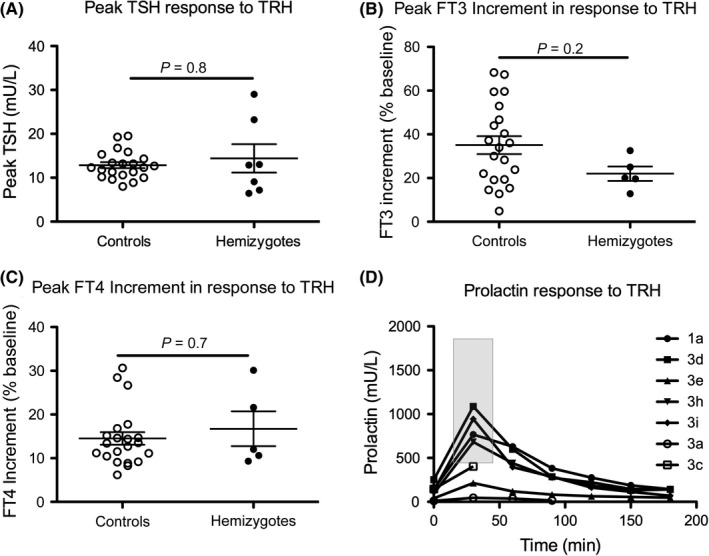
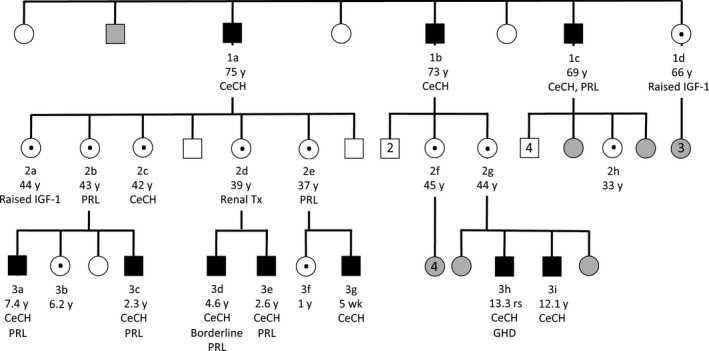
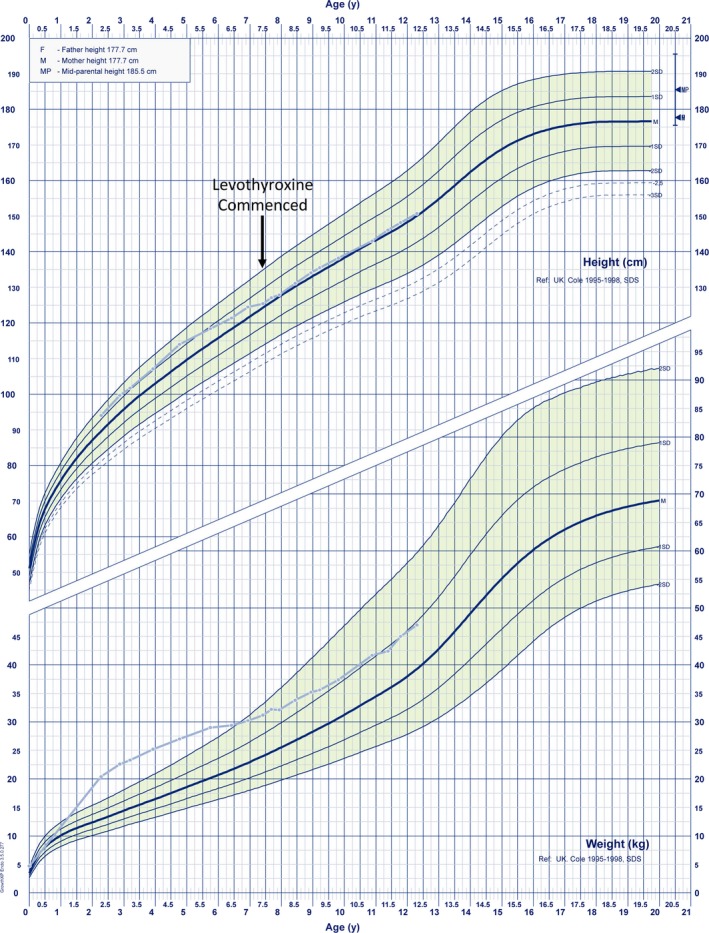
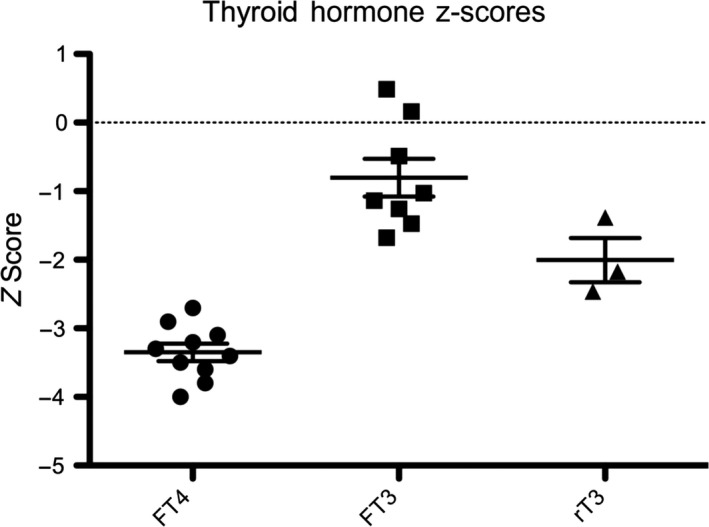
The mutant IGSF1 protein (c.2318T > C, p.L773P) exhibited decreased plasma membrane expression in vitro due to impaired trafficking from the endoplasmic reticulum. Ten hemizygous males and 11 heterozygous females exhibited characteristic endocrine deficits. Ireland operates a TSH-based CH screening programme, which does not detect CeCH; therefore, genetic ascertainment preceded biochemical diagnosis of moderate CH in five of seven boys as well as their 75-year-old grandfather. Clinical features potentially attributable to hypothyroidism were variable; normal free T3 (FT3) and low/low normal reverse T3 (rT3) concentrations suggested that preferential deiodination of FT4 to FT3 may help maintain tissue euthyroidism in some individuals. However, neonatal jaundice, delayed speech or growth, and obesity were observed in seven subjects in whom diagnosis was delayed.
As observed with other IGSF1 mutations, p.L773P results in variably penetrant IGSF1 deficiency syndrome. Our observations emphasize the need for multi-generation genetic ascertainment in affected families, especially where TSH-based CH screening programmes may fail to detect CeCH at birth.
IGSF1 的功能丧失突变导致 X 连锁中枢性先天性甲状腺功能减退症(CeCH),可单独发生或与其他垂体激素缺乏相关。家族内的外显率高度可变,少数杂合子女性也会受到影响。我们在一个大型爱尔兰家族中鉴定并描述了一个新的 IGSF1 突变,并研究了其相关表型。
设计、患者和测量方法:通过直接测序在两名患有 CeCH 的兄弟中鉴定出一个新的 IGSF1 突变,并在体外对其功能后果进行了描述。在更广泛的家族中研究了基因型-表型相关性。
突变的 IGSF1 蛋白(c.2318T>C,p.L773P)由于内质网转运受损而表现出体外血浆膜表达减少。10 名半合子男性和 11 名杂合子女性表现出特征性的内分泌缺陷。爱尔兰实行基于 TSH 的 CH 筛查计划,但不能检测到 CeCH;因此,在七个男孩中的五个以及他们 75 岁的祖父中,遗传确定先于生化诊断中度 CH。可能归因于甲状腺功能减退的临床特征各不相同;正常游离 T3(FT3)和低/正常低反转 T3(rT3)浓度表明,FT4 向 FT3 的优先脱碘可能有助于维持一些个体的组织甲状腺功能正常。然而,在七个诊断延迟的患者中观察到新生儿黄疸、言语或生长迟缓以及肥胖。
与其他 IGSF1 突变一样,p.L773P 导致 IGSF1 缺陷综合征的外显率不同。我们的观察结果强调了在受影响的家族中进行多代遗传确定的必要性,尤其是在 TSH 为基础的 CH 筛查计划可能无法在出生时检测到 CeCH 的情况下。