Arefzadeh Alireza, Khalighinejad Pooyan, Ataeinia Bahar, Parvar Pegah
Endocrinology Department, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
School of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran.
Endocrinol Diabetes Metab Case Rep. 2018 Jul 21;2018. doi: 10.1530/EDM-18-0068. eCollection 2018.
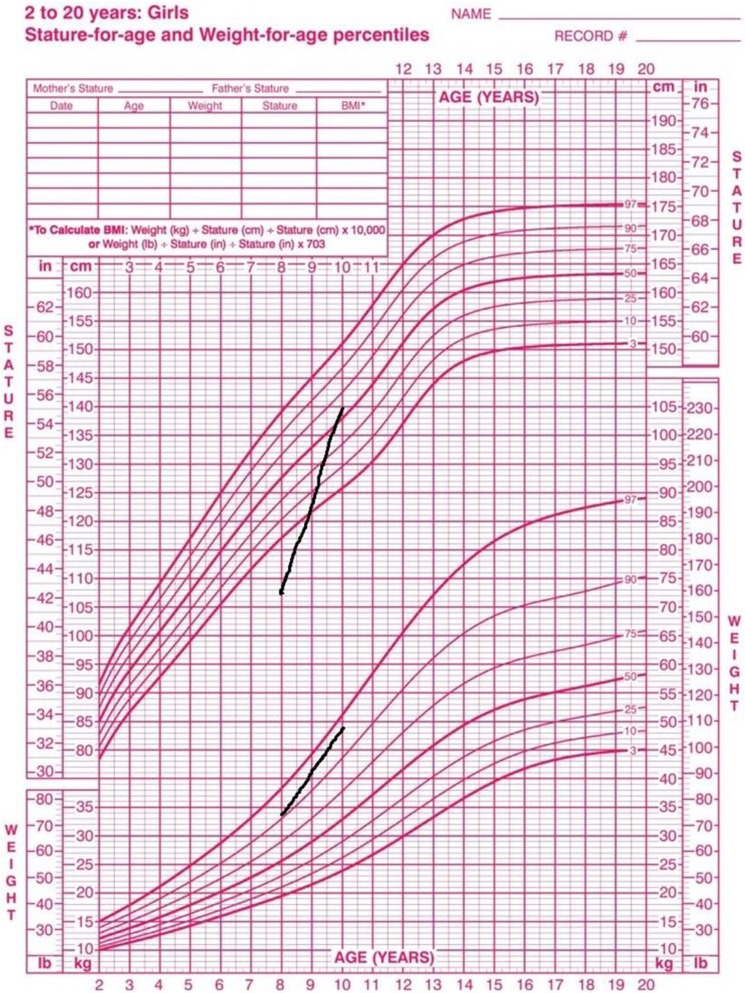
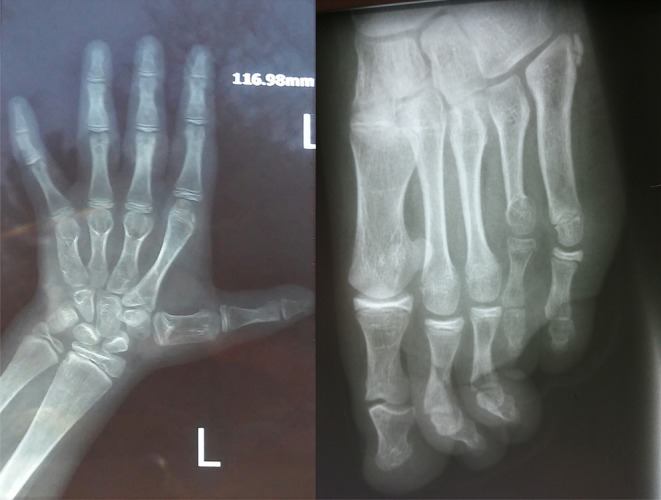
Deletion of chromosome 2q37 results in a rare congenital syndrome known as brachydactyly mental retardation (BDMR) syndrome; a syndrome which has phenotypes similar to Albright hereditary osteodystrophy (AHO) syndrome. In this report, we describe a patient with AHO due to microdeletion in long arm of chromosome 2 [del(2)(q37.3)] who had growth hormone (GH) deficiency, which is a unique feature among reported BDMR cases. This case was presented with shortening of the fourth and fifth metacarpals which along with AHO phenotype, brings pseudopseudohypoparathyroidism (PPHP) and pseudohypoparathyroidism type Ia (PHP-Ia) to mind; however, a genetic study revealed del(2)(q37.3). We recommend clinicians to take BDMR in consideration when they are faced with the features of AHO; although this syndrome is a rare disease, it should be ruled out while diagnosing PPHP or PHP-Ia. Moreover, we recommend evaluation of IGF 1 level and GH stimulation test in patients with BDMR whose height is below the 3rd percentile.
Clinicians must have brachydactyly mental retardation (BDMR) syndrome in consideration when they are faced with the features of Albright hereditary osteodystrophy.Although BDMR syndrome is a rare disease, it should be ruled out while diagnosing PPHP or PHP-Ia.Evaluation of IGF1 level in patients diagnosed with BDMR whose height is below the 3rd percentile is important.
2号染色体q37缺失会导致一种罕见的先天性综合征,即短指智力发育迟缓(BDMR)综合征;该综合征具有与奥尔布赖特遗传性骨营养不良(AHO)综合征相似的表型。在本报告中,我们描述了一名因2号染色体长臂微缺失[del(2)(q37.3)]而患有AHO的患者,其存在生长激素(GH)缺乏,这是已报道的BDMR病例中的独特特征。该病例表现为第四和第五掌骨缩短,连同AHO表型,让人联想到假假性甲状旁腺功能减退症(PPHP)和Ia型假性甲状旁腺功能减退症(PHP-Ia);然而,基因研究显示为del(2)(q37.3)。我们建议临床医生在面对AHO特征时考虑BDMR;尽管该综合征是一种罕见疾病,但在诊断PPHP或PHP-Ia时应排除。此外,我们建议对身高低于第3百分位数的BDMR患者进行胰岛素样生长因子1(IGF 1)水平评估和GH刺激试验。
临床医生在面对奥尔布赖特遗传性骨营养不良特征时必须考虑短指智力发育迟缓(BDMR)综合征。尽管BDMR综合征是一种罕见疾病,但在诊断PPHP或PHP-Ia时应排除。对身高低于第3百分位数的BDMR确诊患者进行IGF1水平评估很重要。