Xiao Haopeng, Hwang Ju Eun, Wu Ronghu
School of Chemistry and Biochemistry and the Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, Georgia 30332, USA.
Int J Mass Spectrom. 2018 Jun;429:66-75. doi: 10.1016/j.ijms.2017.05.010. Epub 2017 May 27.
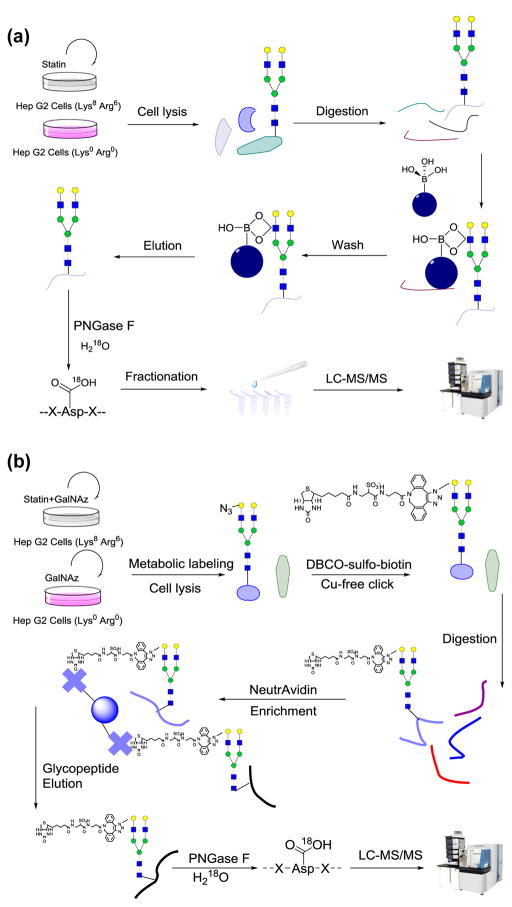
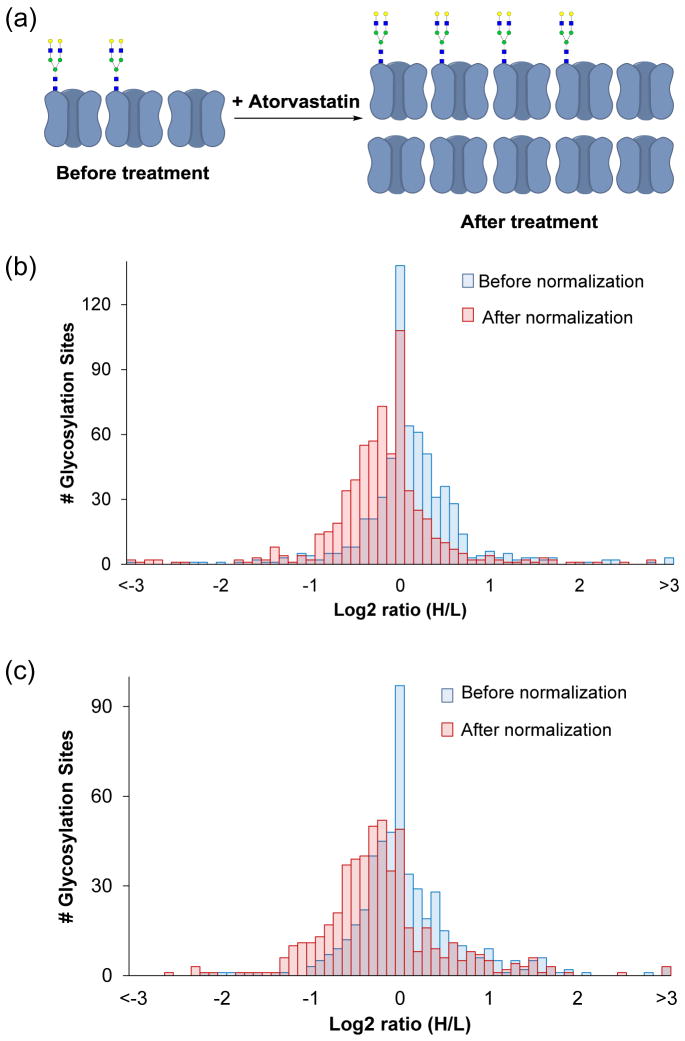
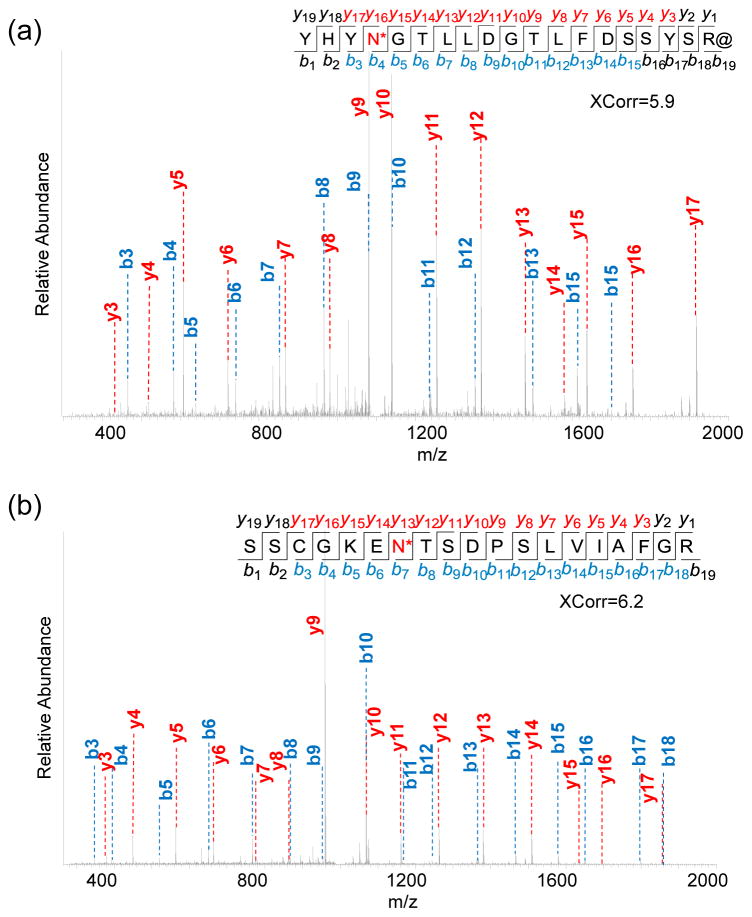
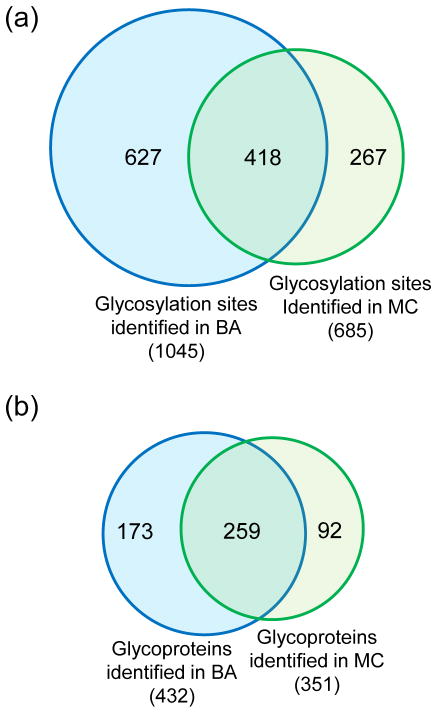
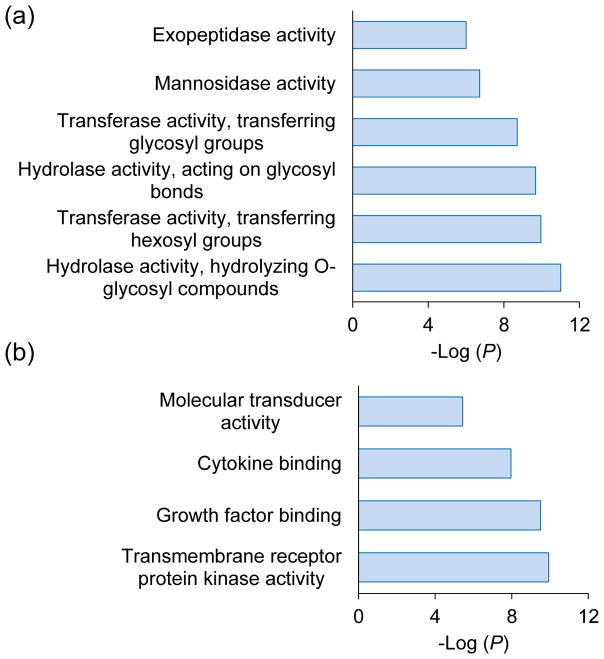
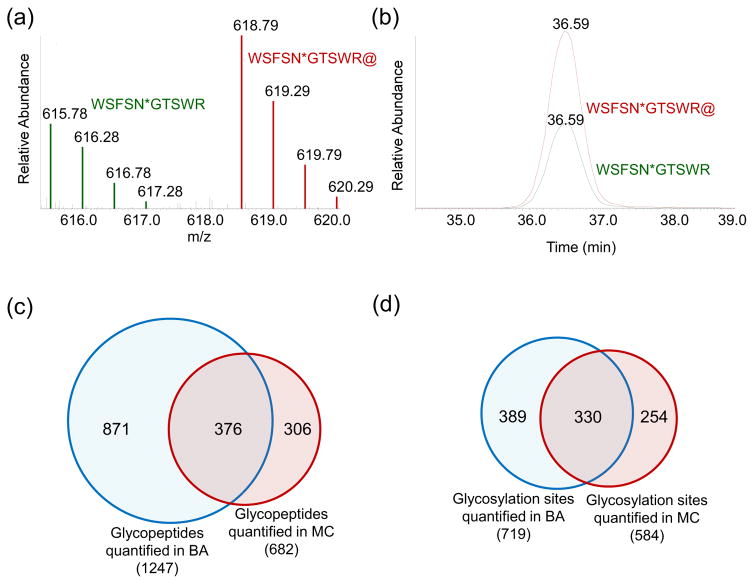
Protein N-glycosylation is essential for mammalian cell survival and is well-known to be involved in many biological processes. Aberrant glycosylation is directly related to human disease including cancer and infectious diseases. Global analysis of protein N-glycosylation will allow a better understanding of protein functions and cellular activities. Mass spectrometry (MS)-based proteomics provides a unique opportunity to site-specifically characterize protein glycosylation on a large scale. Due to the complexity of biological samples, effective enrichment methods are critical prior to MS analysis. Here, we compared two lectin-independent methods to enrich glycopeptides for the global analysis of protein N-glycosylation by MS. The first boronic acid-based enrichment (BA) method benefits from the universal and reversible interactions between boronic acid and sugars; the other method utilizes metabolic labeling and click chemistry (MC) to incorporate a chemical handle into glycoproteins for future affinity enrichment. We comprehensively compared the performance of the two methods in the identification and quantification of glycoproteins in statin-treated liver cells. Based on the current results, the BA method is more universal in enriching glycopeptides, while with the MC method, cell surface glycoproteins were highly enriched, and the quantification results appear to be more dynamic because only the newly-synthesized glycoproteins were analyzed. In addition, we normalized the glycosylation site ratios by the corresponding parent protein ratios to reflect the real modification changes. In combination with MS-based proteomics, effective enrichment methods will vertically advance protein glycosylation research.
蛋白质N-糖基化对于哺乳动物细胞的存活至关重要,并且众所周知它参与许多生物过程。异常糖基化与包括癌症和传染病在内的人类疾病直接相关。对蛋白质N-糖基化进行全局分析将有助于更好地理解蛋白质功能和细胞活动。基于质谱(MS)的蛋白质组学为大规模地对蛋白质糖基化进行位点特异性表征提供了独特的机会。由于生物样品的复杂性,在进行MS分析之前,有效的富集方法至关重要。在此,我们比较了两种不依赖凝集素的方法,用于富集糖肽以通过MS对蛋白质N-糖基化进行全局分析。第一种基于硼酸的富集(BA)方法受益于硼酸与糖之间普遍且可逆的相互作用;另一种方法利用代谢标记和点击化学(MC)将化学手柄掺入糖蛋白中,以便将来进行亲和富集。我们全面比较了这两种方法在他汀类药物处理的肝细胞中糖蛋白鉴定和定量方面的性能。基于目前的结果,BA方法在富集糖肽方面更具通用性,而对于MC方法,细胞表面糖蛋白高度富集,并且定量结果似乎更具动态性,因为仅分析了新合成的糖蛋白。此外,我们通过相应的亲本蛋白质比率对糖基化位点比率进行归一化,以反映实际的修饰变化。结合基于MS的蛋白质组学,有效的富集方法将在垂直方向上推动蛋白质糖基化研究。