Joint Institute for Food Safety and Applied Nutrition, Center for Food Safety and Security Systems, University of Maryland, College Park, MD, United States of America.
Department of Diagnostic Medicine/Pathobiology, College of Veterinary Medicine, Kansas State University, Manhattan, KS, United States of America.
PLoS One. 2018 Aug 28;13(8):e0202775. doi: 10.1371/journal.pone.0202775. eCollection 2018.
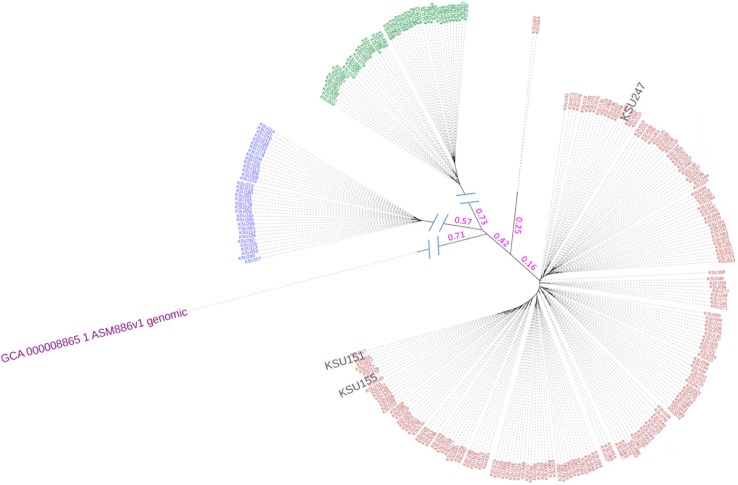
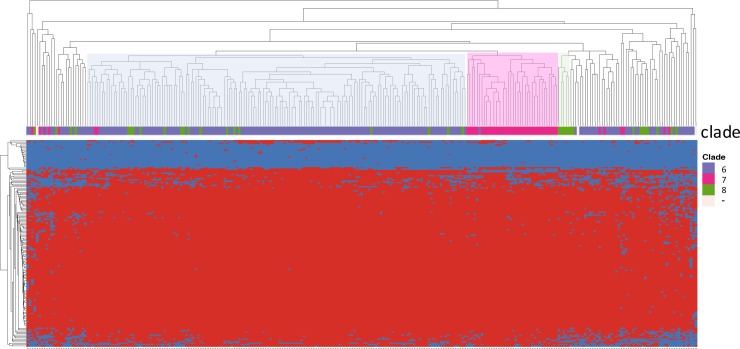
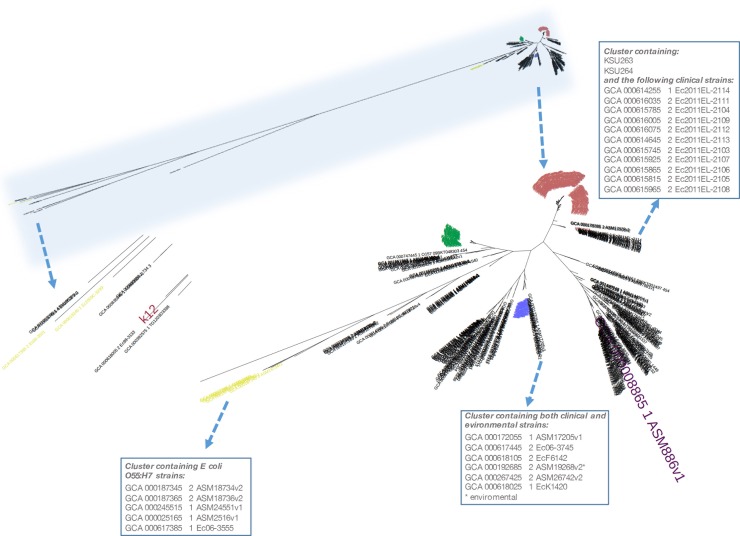
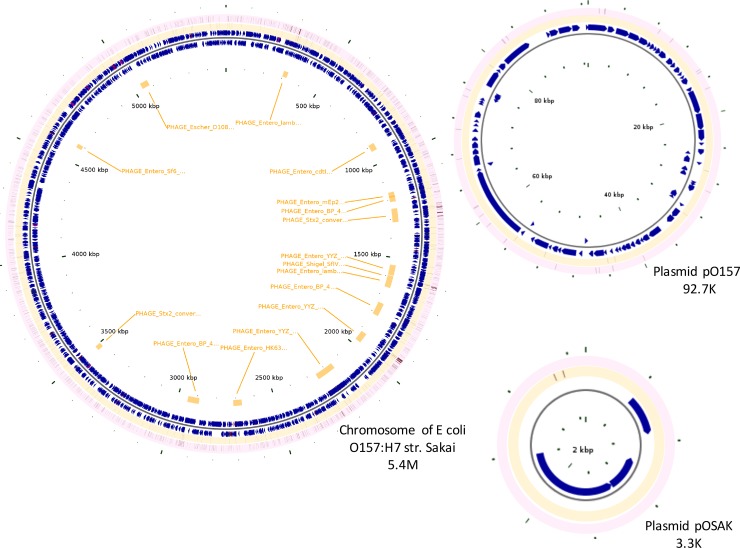
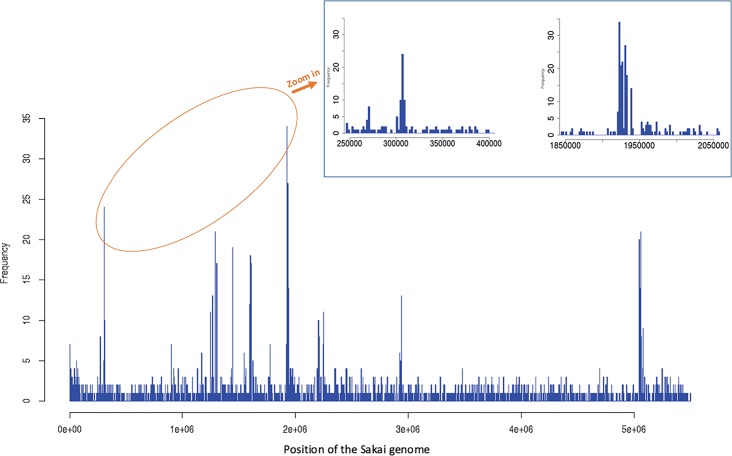
Escherichia coli serotype O157:H7 continues to pose a serious health threat to human beings. Cattle, a major reservoir of the pathogen, harbor E. coli O157:H7 in their gastrointestinal tract and shed variable concentrations of E. coli O157:H7 into the environment. Genetic characterization of cattle-shed E. coli O157 strains is of interest to the livestock industry, food business, and public health community. The present study applied whole genome shotgun sequencing (WGS) and single nucleotide variant (SNV) calling to characterize 279 cattle-shed E. coli O157:H7 strains isolated from a single feedlot located in southwestern region of the US. More than 4,000 SNVs were identified among the strains and the resultant phylogenomic tree revealed three major groups. Using the Sakai strain genome as reference, more than 2,000 SNVs were annotated and a detailed SNV map generated. Results clearly revealed highly polymorphic loci along the E. coli O157:H7 genome that aligned with the prophage regions and highly variant genes involved in processing bacterial genetic information. The WGS data were further profiled against a comprehensive virulence factor database (VFDB) for virulence gene identification. Among the total 285 virulence genes identified, only 132 were present in all the strains. There were six virulence genes unique to single isolates. Our findings suggested that the genome variations of the E. coli O157:H7 were mainly attributable to dynamics of certain phages, and the bacterial strains have variable virulence gene profiles, even though they came from a single cattle population, which may explain the differences in pathogenicity, host prevalence, and transmissibility by E. coli O157:H7.
产志贺毒素大肠杆菌 O157:H7 持续对人类健康构成严重威胁。牛是该病原体的主要宿主,其胃肠道中携带产志贺毒素大肠杆菌 O157:H7,并将不同浓度的产志贺毒素大肠杆菌 O157:H7 排入环境中。牛源产志贺毒素大肠杆菌 O157 菌株的遗传特征是畜牧业、食品企业和公共卫生界关注的焦点。本研究应用全基因组鸟枪法测序(WGS)和单核苷酸变异(SNV)调用技术,对来自美国西南部单一饲养场的 279 株牛源产志贺毒素大肠杆菌 O157:H7 进行了特征描述。在这些菌株中鉴定出超过 4000 个 SNV,由此产生的系统发育树显示出三个主要群组。使用坂井氏菌株基因组作为参考,对超过 2000 个 SNV 进行了注释,并生成了详细的 SNV 图谱。结果清楚地揭示了大肠杆菌 O157:H7 基因组中高度多态性的基因座,这些基因座与噬菌体区域和涉及处理细菌遗传信息的高度变异基因对齐。进一步将 WGS 数据与综合毒力因子数据库(VFDB)进行对比,以鉴定毒力基因。在所鉴定的 285 个毒力基因中,只有 132 个存在于所有菌株中。有 6 个毒力基因仅存在于单个分离株中。我们的研究结果表明,大肠杆菌 O157:H7 的基因组变异主要归因于某些噬菌体的动态变化,而且细菌株具有不同的毒力基因谱,即使它们来自单一的牛群,这可能解释了产志贺毒素大肠杆菌 O157:H7 致病性、宿主流行率和传染性的差异。