Universidade Nove de Julho, Rua Vergueiro 249, 01504-001 São Paulo, SP, Brazil.
Universidade Federal de São Paulo, Rua Pedro de Toledo 697, 04039-001 São Paulo, SP, Brazil.
Oxid Med Cell Longev. 2018 Aug 6;2018:6736721. doi: 10.1155/2018/6736721. eCollection 2018.
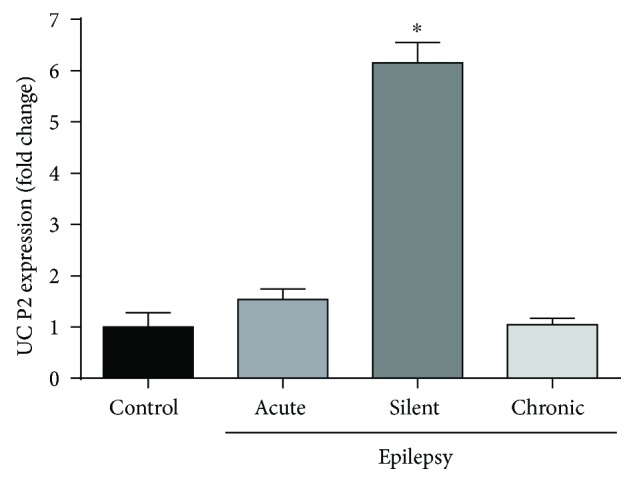
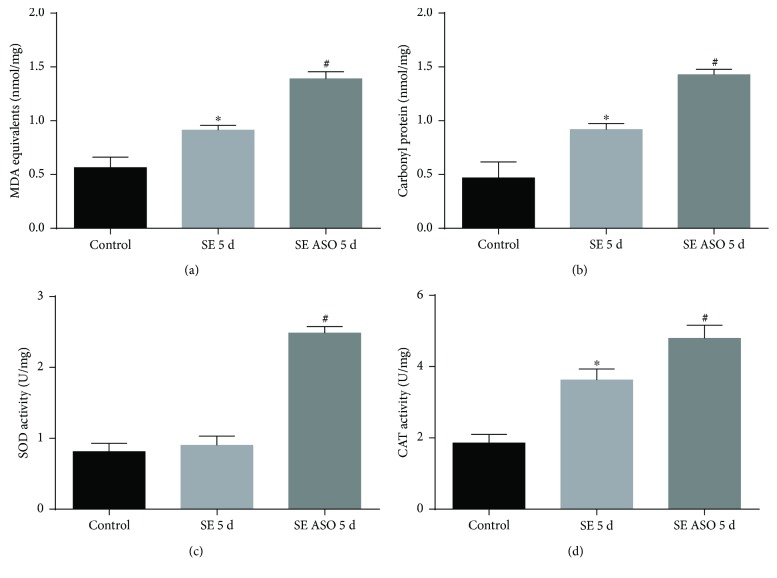
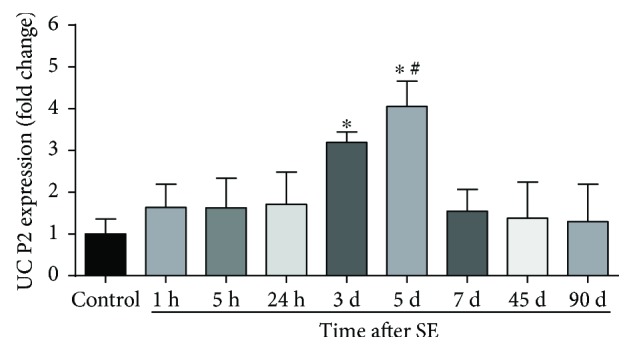
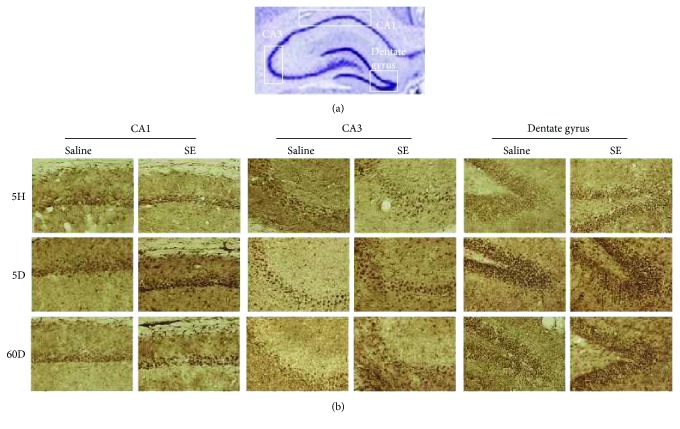
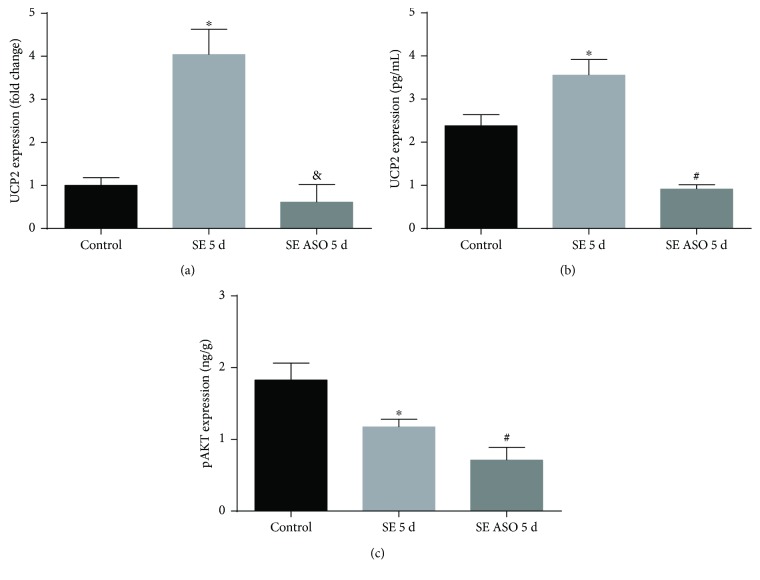
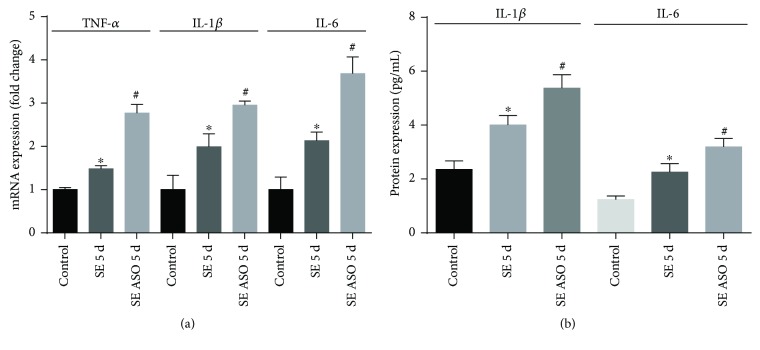
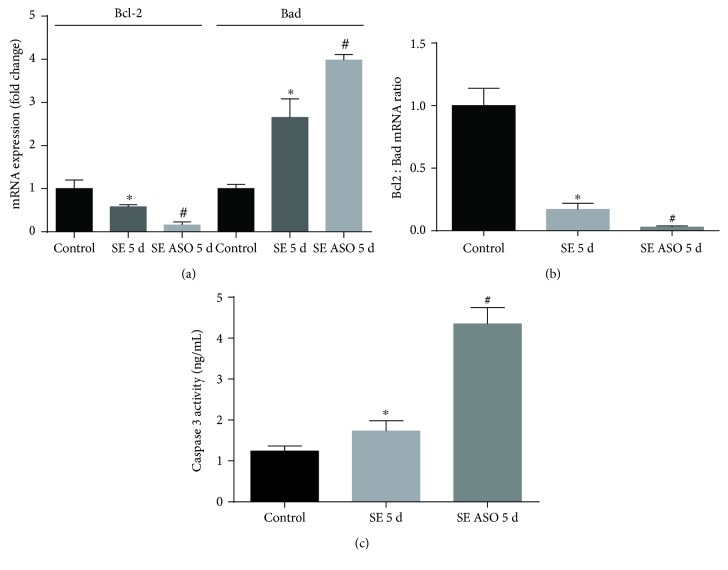
Neuroprotection is a desirable process in many neurological disorders, yet complex mechanisms involved in this field are not completely understood. The pilocarpine epilepsy model causes potent, seizure-induced excitotoxicity cell death and mitochondria impairment. The present study is aimed at investigating the role of UCP2, a ROS negative regulator, in the neuroprotection after cholinergic insult. Our data demonstrated that UCP2 expression was augmented in the rat hippocampus 3 days after (SE), reaching a peak on the fifth day, then returning to basal levels. Concomitantly, phospho-AKT expression levels were higher in the hippocampus during the early silent phase (5 days after SE). Additionally, it was demonstrated that the blockade of UCP2 by antisense oligonucleotides (ASO) in SE rats successfully diminished both UCP2 mRNA and protein contents. SE ASO rats presented increased mitochondrial proapoptotic factor expression, caspase-3 activity, inflammatory cytokine expression, and ROS formation. Moreover, ASO treatment diminished p-AKT expression and antioxidant enzyme activities after pilocarpine insult. In conclusion, the present results highlight the neuroprotective actions of UCP2, acting in the inhibition of apoptotic factors and oxidative stress, to increase neuron survival after SE onset.
神经保护是许多神经紊乱疾病中所期望的过程,但该领域涉及的复杂机制尚未完全被理解。匹罗卡品癫痫模型可导致强烈的、由癫痫发作引起的兴奋性毒性细胞死亡和线粒体损伤。本研究旨在探讨 UCP2(一种 ROS 负调节剂)在胆碱能损伤后的神经保护中的作用。我们的数据表明,UCP2 表达在 SE 后 3 天(SE)时在大鼠海马体中增强,在第 5 天达到峰值,然后恢复到基础水平。同时,在 SE 后的早期沉默期(SE 后 5 天),磷酸化 AKT 表达水平在海马体中更高。此外,在 SE 大鼠中用反义寡核苷酸(ASO)阻断 UCP2 成功地降低了 UCP2 mRNA 和蛋白质含量。SE ASO 大鼠表现出增加的线粒体促凋亡因子表达、caspase-3 活性、炎症细胞因子表达和 ROS 形成。此外,ASO 处理在匹罗卡品损伤后降低了 p-AKT 表达和抗氧化酶活性。总之,本研究结果强调了 UCP2 的神经保护作用,通过抑制凋亡因子和氧化应激,增加 SE 发作后神经元的存活。