Mitochondria and Metabolism Center, Department of Anesthesiology and Pain Medicine, University of Washington, Seattle, WA 98109, USA; Department of Pathology, University of Washington, Seattle, WA, 98195, USA.
Mitochondria and Metabolism Center, Department of Anesthesiology and Pain Medicine, University of Washington, Seattle, WA 98109, USA.
J Mol Cell Cardiol. 2018 Oct;123:38-45. doi: 10.1016/j.yjmcc.2018.08.022. Epub 2018 Aug 27.
Ischemic heart disease (IHD) is a leading cause of mortality. The most effective intervention for IHD is reperfusion, which ironically causes ischemia reperfusion (I/R) injury mainly due to oxidative stress-induced cardiomyocyte death. The exact mechanism and site of reactive oxygen species (ROS) generation during I/R injury remain elusive.
We aim to test the hypothesis that Complex I-mediated forward and reverse electron flows are the major source of ROS in I/R injury of the heart.
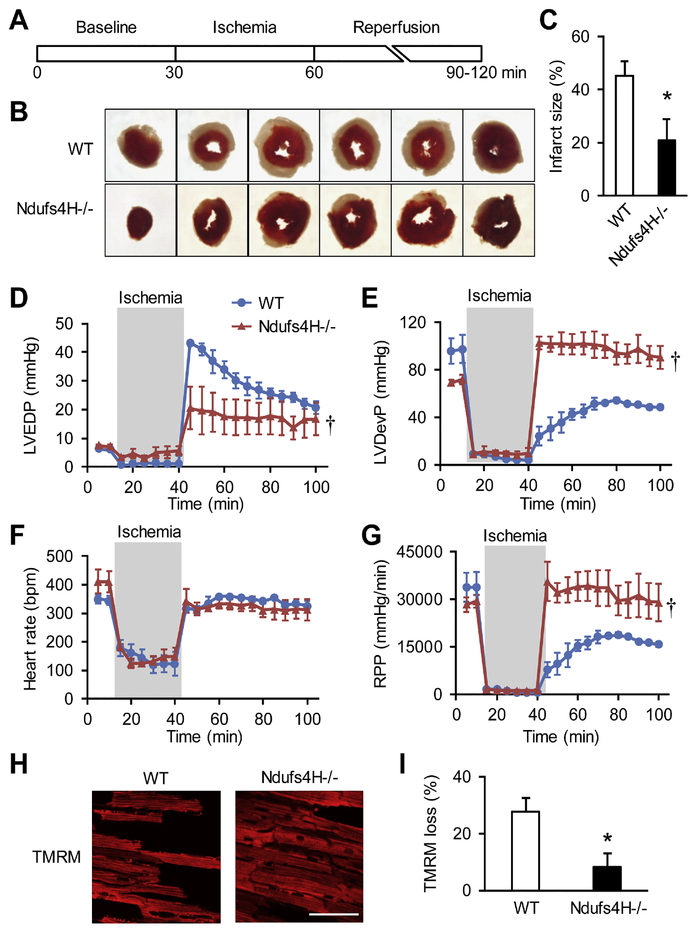
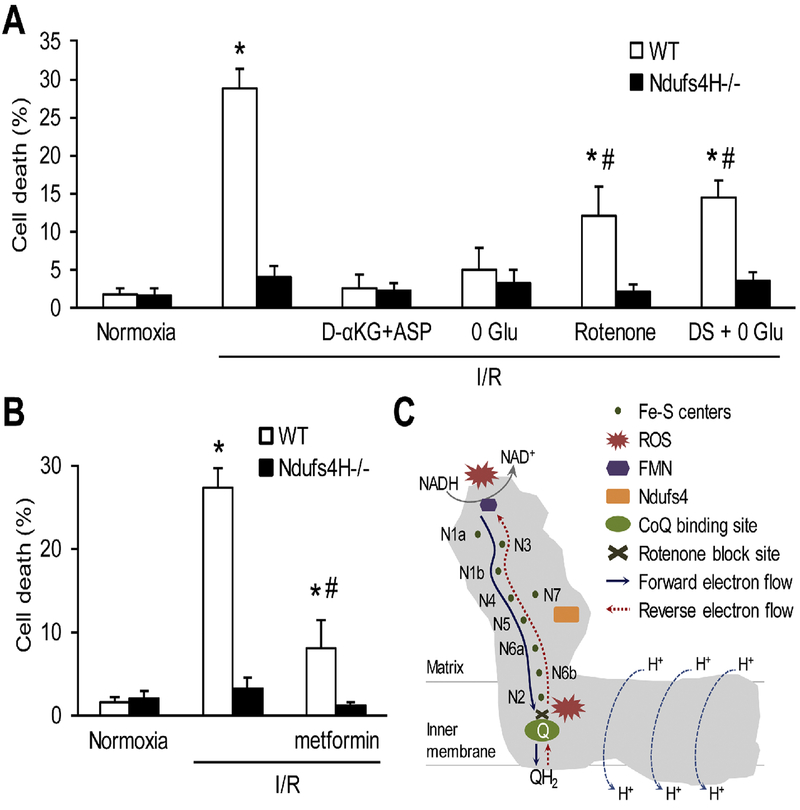
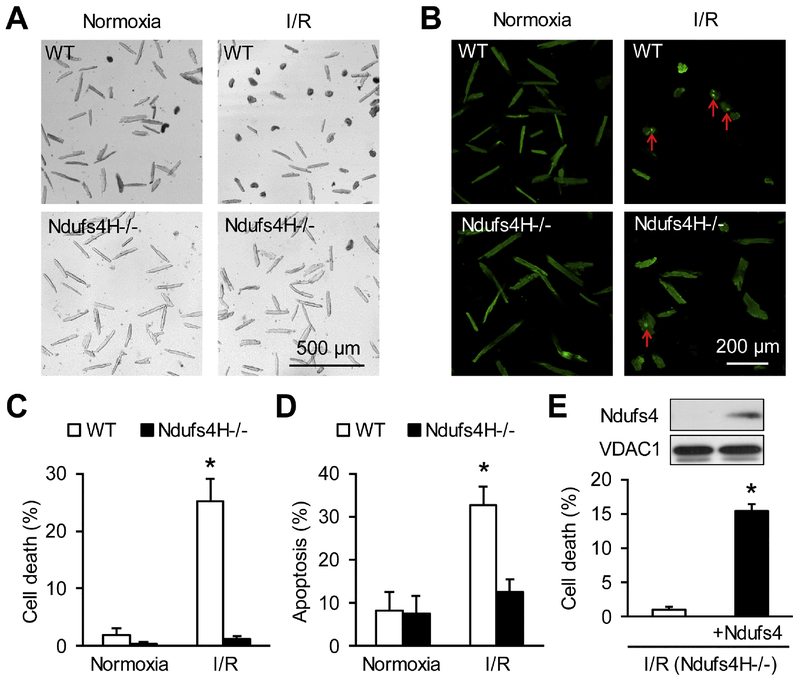
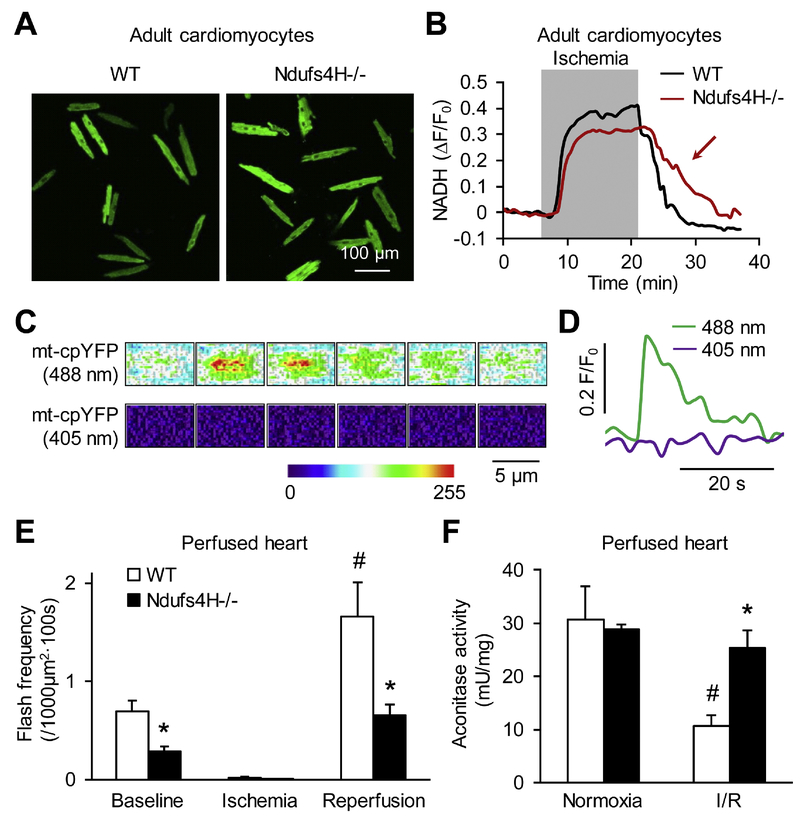
We used a genetic model of mitochondrial Complex I deficiency, in which a Complex I assembling subunit, Ndufs4 was knocked out in the heart (Ndufs4H-/-). The Langendorff perfused Ndufs4H-/- hearts exhibited significantly reduced infarct size (45.3 ± 5.5% in wild type vs 20.9 ± 8.1% in Ndufs4H-/-), recovered contractile function, and maintained mitochondrial membrane potential after no flow ischemia and subsequent reperfusion. In cultured adult cardiomyocytes from Ndufs4H-/- mice, I/R mimetic treatments caused minimal cell death. Reintroducing Ndufs4 in Ndufs4H-/- cardiomyocytes abolished the protection. Mitochondrial NADH declined much slower in Ndufs4H-/- cardiomyocytes during reperfusion suggesting decreased forward electron flow. Mitochondrial flashes, a marker for mitochondrial respiration, were inhibited in Ndufs4H-/- cardiomyocytes at baseline and during I/R, which was accompanied by preserved aconitase activity suggesting lack of oxidative damage. Finally, pharmacological blockade of forward and reverse electron flow at Complex I inhibited I/R-induced cell death.
These results provide the first genetic evidence supporting the central role of mitochondrial Complex I in I/R injury of mouse heart. The study also suggests that both forward and reverse electron flows underlie oxidative cardiomyocyte death during reperfusion.
缺血性心脏病(IHD)是主要的死亡原因。IHD 最有效的干预措施是再灌注,但再灌注会引起缺血再灌注(I/R)损伤,主要是由于氧化应激诱导的心肌细胞死亡。在 I/R 损伤期间,活性氧(ROS)生成的确切机制和部位仍不清楚。
我们旨在检验以下假设,即复合物 I 介导的正向和逆向电子流是心脏 I/R 损伤中 ROS 的主要来源。
我们使用了一种线粒体复合物 I 缺陷的遗传模型,其中一种复合物 I 组装亚基 Ndufs4 在心脏中被敲除(Ndufs4H-/-)。Langendorff 灌注的 Ndufs4H-/-心脏的梗死面积明显减小(野生型为 45.3±5.5%,Ndufs4H-/-为 20.9±8.1%),在无血流缺血和随后的再灌注后,收缩功能得到恢复,并且线粒体膜电位得以维持。在 Ndufs4H-/-小鼠的成年心肌细胞中,I/R 模拟处理引起的细胞死亡最小。在 Ndufs4H-/-心肌细胞中重新引入 Ndufs4 则消除了这种保护作用。在再灌注过程中,Ndufs4H-/-心肌细胞中的线粒体 NADH 下降速度要慢得多,这表明正向电子流减少。Ndufs4H-/-心肌细胞在基线和 I/R 期间的线粒体闪烁(一种线粒体呼吸的标志物)受到抑制,同时伴有无氧化损伤的 aconitase 活性的保留。最后,复合物 I 上正向和逆向电子流的药理学阻断抑制了 I/R 诱导的细胞死亡。
这些结果提供了第一个支持线粒体复合物 I 在小鼠心脏 I/R 损伤中的核心作用的遗传证据。该研究还表明,在再灌注期间,正向和逆向电子流都导致氧化应激诱导的心肌细胞死亡。