Montenegro Renan Magalhães, Costa-Riquetto Aline Dantas, Fernandes Virgínia Oliveira, Montenegro Ana Paula Dias Rangel, de Santana Lucas Santos, Jorge Alexander Augusto de Lima, Karbage Lia Beatriz de Azevedo Souza, Aguiar Lindenberg Barbosa, Carvalho Francisco Herlânio Costa, Teles Milena Gurgel, d'Alva Catarina Brasil
Brazilian Group for the Study of Inherited and Acquired Lipodystrophies, Faculdade de Medicina, Universidade Federal do Ceará, Fortaleza, Brazil.
Monogenic Diabetes Group, Genetic Endocrinology Unit (LIM25), Hospital das Clinicas da Faculdade de Medicina, Universidade de São Paulo, São Paulo, Brazil.
Front Endocrinol (Lausanne). 2018 Aug 20;9:458. doi: 10.3389/fendo.2018.00458. eCollection 2018.
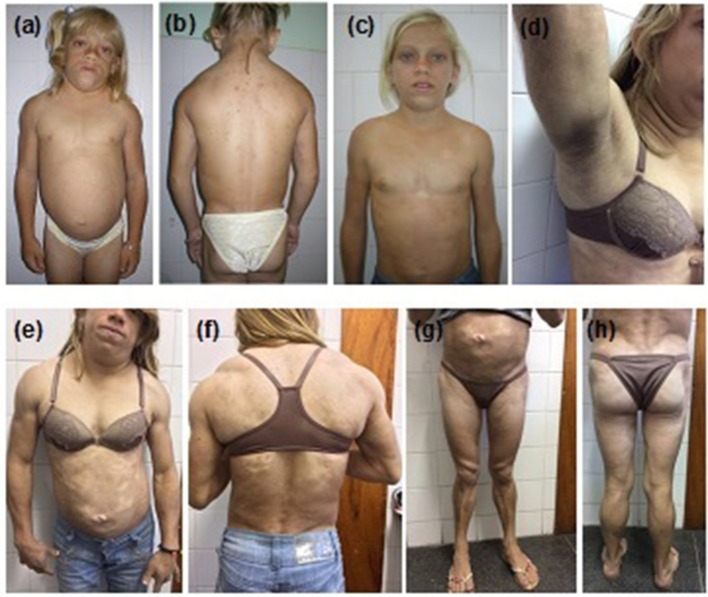
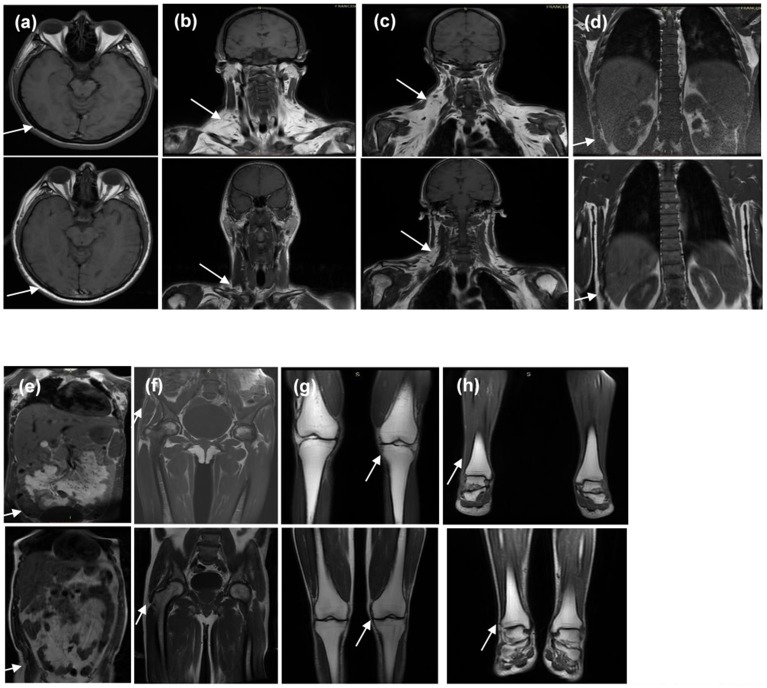
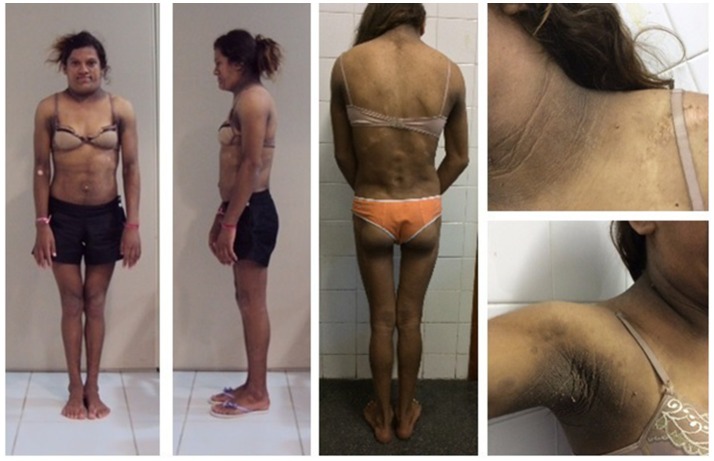
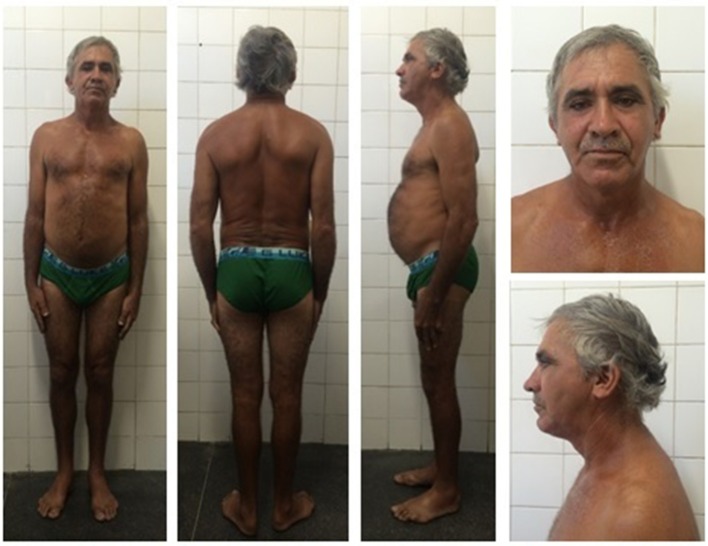
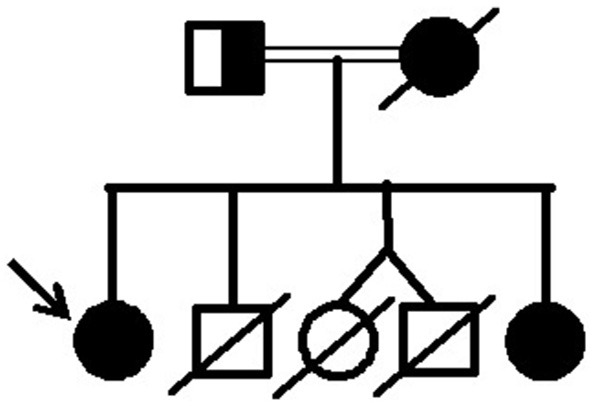
Dunnigan-type familial partial lipodystrophy (FPLD2) is a rare autosomal dominant disease caused by heterozygous mutations in the gene that results in regional loss of subcutaneous adipose tissue with onset in puberty. However, a generalized lipodystrophy phenotype has also been associated with heterozygous mutations in this gene, demonstrating the noticeable phenotypic heterogeneity of this disease. We report and describe clinical and metabolic features of four patients from the same family with the p.R582C mutation, three homozygous and one in the heterozygous state that present with three distinct lipodystrophic phenotypes. Case description: The proband was a 12-year-old girl who developed severe subcutaneous fat atrophy in limbs and abdomen followed by a remarkable dorsocervical fat accumulation in adulthood along with diabetes at age 23. The proband's sister was a phenotypically normal girl who developed hypertriglyceridemia at age 8, progressive features of partial lipodystrophy at age 11, and diabetes at age 22. The proband's mother was first examined at age 32, presenting diabetes and a severe generalized lipodystrophic phenotype; she developed kidney failure at age 41 and died due to diabetic complications. The proband's father was a 50-year-old man with abdominal fat concentration that was initially considered phenotypically normal. Massively parallel sequencing using a platform of genes related to genetic lipodystrophies, followed by Sanger sequencing, revealed the transversion c.1744C>T at exon 11 of the gene (p.R582C) in the homozygous (mother and daughters) and heterozygous (father) states. We documented three distinct phenotypes of the homozygous and heterozygous p. R582C mutation in the same kindred, illustrating that FPLD2 linked to mutations in this gene is a disease of great clinical heterogeneity, possibly due to associated environmental or genetic factors.
邓尼根型家族性部分脂肪营养不良(FPLD2)是一种罕见的常染色体显性疾病,由该基因的杂合突变引起,导致青春期开始时皮下脂肪组织区域性缺失。然而,该基因的杂合突变也与全身性脂肪营养不良表型相关,表明这种疾病具有显著的表型异质性。我们报告并描述了来自同一家族的四名携带p.R582C突变患者的临床和代谢特征,其中三名纯合子和一名杂合子患者呈现出三种不同的脂肪营养不良表型。病例描述:先证者是一名12岁女孩,四肢和腹部出现严重的皮下脂肪萎缩,成年后颈背部脂肪显著堆积,并在23岁时患上糖尿病。先证者的妹妹是一名表型正常的女孩,8岁时出现高甘油三酯血症,11岁时出现部分脂肪营养不良的进行性特征,22岁时患上糖尿病。先证者的母亲首次检查时32岁,患有糖尿病和严重的全身性脂肪营养不良表型;她在41岁时出现肾衰竭,因糖尿病并发症死亡。先证者的父亲是一名50岁男性,腹部脂肪集中,最初被认为表型正常。使用与遗传性脂肪营养不良相关的基因平台进行大规模平行测序,随后进行桑格测序,发现在该基因第11外显子处存在纯合(母亲和女儿)和杂合(父亲)状态的颠换c.1744C>T(p.R582C)。我们记录了同一家族中纯合和杂合p.R582C突变的三种不同表型,说明与该基因突变相关的FPLD2是一种临床异质性很大的疾病,可能是由于相关的环境或遗传因素所致。