Environmental Chemistry and Technology Program, University of Wisconsin-Madison, Madison, Wisconsin, USA
Department of Bacteriology, University of Wisconsin-Madison, Madison, Wisconsin, USA.
mSphere. 2018 Sep 5;3(5):e00327-18. doi: 10.1128/mSphere.00327-18.
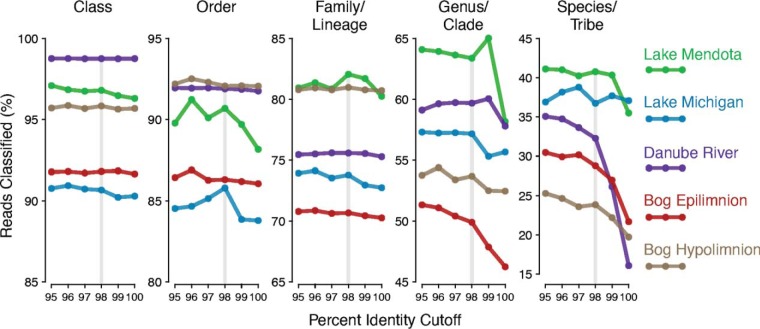
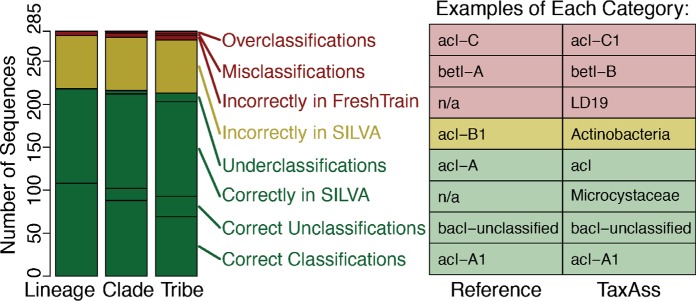
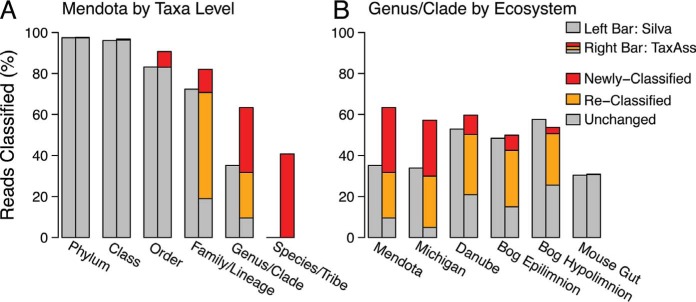
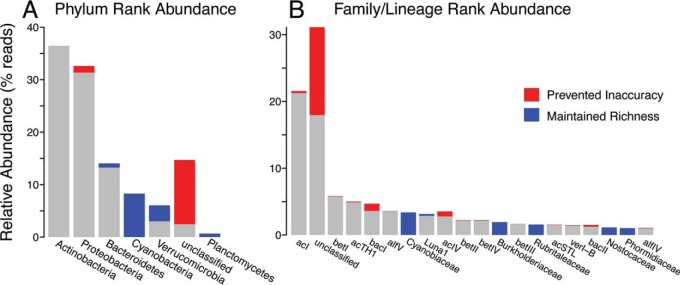
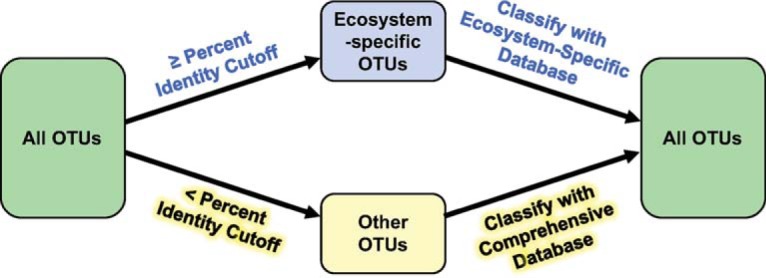
Taxonomy assignment of freshwater microbial communities is limited by the minimally curated phylogenies used for large taxonomy databases. Here we introduce TaxAss, a taxonomy assignment workflow that classifies 16S rRNA gene amplicon data using two taxonomy reference databases: a large comprehensive database and a small ecosystem-specific database rigorously curated by scientists within a field. We applied TaxAss to five different freshwater data sets using the comprehensive SILVA database and the freshwater-specific FreshTrain database. TaxAss increased the percentage of the data set classified compared to using only SILVA, especially at fine-resolution family to species taxon levels, while across the freshwater test data sets classifications increased by as much as 11 to 40% of total reads. A similar increase in classifications was not observed in a control mouse gut data set, which was not expected to contain freshwater bacteria. TaxAss also maintained taxonomic richness compared to using only the FreshTrain across all taxon levels from phylum to species. Without TaxAss, most organisms not represented in the FreshTrain were unclassified, but at fine taxon levels, incorrect classifications became significant. We validated TaxAss using simulated amplicon data derived from full-length clone libraries and found that 96 to 99% of test sequences were correctly classified at fine resolution. TaxAss splits a data set's sequences into two groups based on their percent identity to reference sequences in the ecosystem-specific database. Sequences with high similarity to sequences in the ecosystem-specific database are classified using that database, and the others are classified using the comprehensive database. TaxAss is free and open source and is available at https://www.github.com/McMahonLab/TaxAss Microbial communities drive ecosystem processes, but microbial community composition analyses using 16S rRNA gene amplicon data sets are limited by the lack of fine-resolution taxonomy classifications. Coarse taxonomic groupings at the phylum, class, and order levels lump ecologically distinct organisms together. To avoid this, many researchers define operational taxonomic units (OTUs) based on clustered sequences, sequence variants, or unique sequences. These fine-resolution groupings are more ecologically relevant, but OTU definitions are data set dependent and cannot be compared between data sets. Microbial ecologists studying freshwater have curated a small, ecosystem-specific taxonomy database to provide consistent and up-to-date terminology. We created TaxAss, a workflow that leverages this database to assign taxonomy. We found that TaxAss improves fine-resolution taxonomic classifications (family, genus, and species). Fine taxonomic groupings are more ecologically relevant, so they provide an alternative to OTU-based analyses that is consistent and comparable between data sets.
淡水微生物群落的分类学分配受到用于大型分类学数据库的最小管理系统发育树的限制。在这里,我们介绍了 TaxAss,这是一种分类学分配工作流程,它使用两个分类学参考数据库对 16S rRNA 基因扩增子数据进行分类:一个大型综合数据库和一个由该领域科学家严格管理的小型生态系统特定数据库。我们使用综合 SILVA 数据库和淡水特定的 FreshTrain 数据库将 TaxAss 应用于五个不同的淡水数据集。与仅使用 SILVA 相比,TaxAss 增加了数据集分类的百分比,特别是在精细分辨率的科到种分类群水平,而在整个淡水测试数据集分类中,增加了多达 11%至 40%的总读数。在一个不期望包含淡水细菌的对照鼠肠数据集上,没有观察到类似的分类增加。TaxAss 还保持了与在所有分类群水平(从门到种)仅使用 FreshTrain 相比的分类丰富度。如果没有 TaxAss,大多数不在 FreshTrain 中表示的生物体都未被分类,但在精细分类群水平上,不正确的分类变得很重要。我们使用源自全长克隆文库的模拟扩增子数据验证了 TaxAss,发现 96%至 99%的测试序列在精细分辨率下得到正确分类。TaxAss 根据其与生态系统特定数据库中参考序列的百分比身份将数据集的序列分为两组。与生态系统特定数据库中的序列具有高相似性的序列使用该数据库进行分类,而其他序列则使用综合数据库进行分类。TaxAss 是免费的开源软件,可在 https://www.github.com/McMahonLab/TaxAss 上获得。微生物群落驱动生态系统过程,但使用 16S rRNA 基因扩增子数据集进行微生物群落组成分析受到缺乏精细分辨率分类学分类的限制。在门、纲和目等粗分类群水平上,将具有生态差异的生物体混在一起。为了避免这种情况,许多研究人员基于聚类序列、序列变体或独特序列定义操作分类单位 (OTU)。这些精细分辨率的分组更具生态相关性,但 OTU 定义是数据集特定的,并且不能在数据集之间进行比较。研究淡水的微生物生态学家已经整理了一个小型的、生态系统特定的分类学数据库,以提供一致的、最新的术语。我们创建了 TaxAss,这是一种利用该数据库进行分类的工作流程。我们发现 TaxAss 提高了精细分辨率的分类学分类(科、属和种)。精细的分类群更具生态相关性,因此它们提供了一种替代基于 OTU 的分析方法,这种方法在数据集之间是一致和可比较的。