Aix Marseille Université, INSERM, UMR-S1076, Vascular Research Center Marseille, Marseille, France.
Department of Hemostasis and Thrombosis, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, USA.
J Thromb Haemost. 2018 Nov;16(11):2322-2335. doi: 10.1111/jth.14291. Epub 2018 Oct 12.
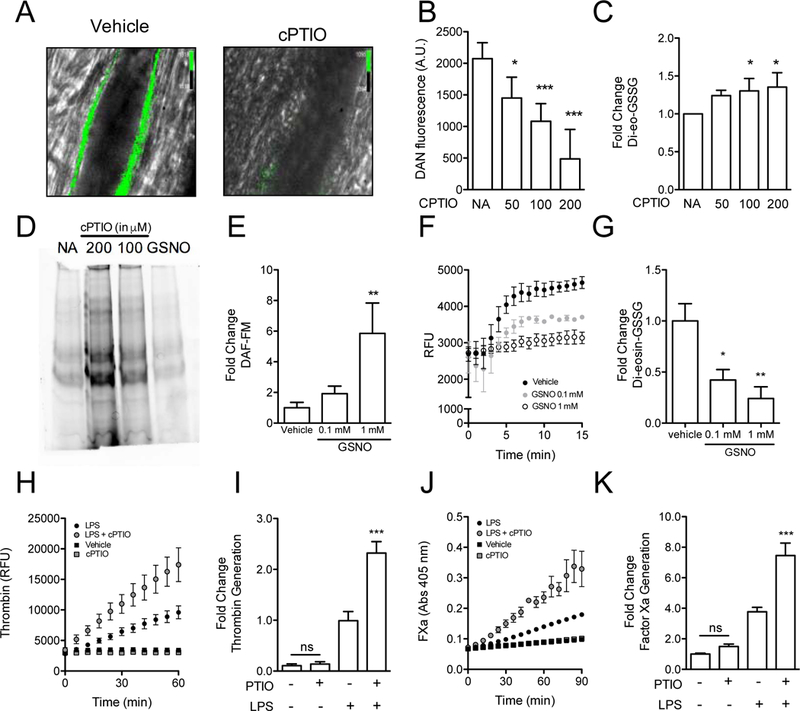
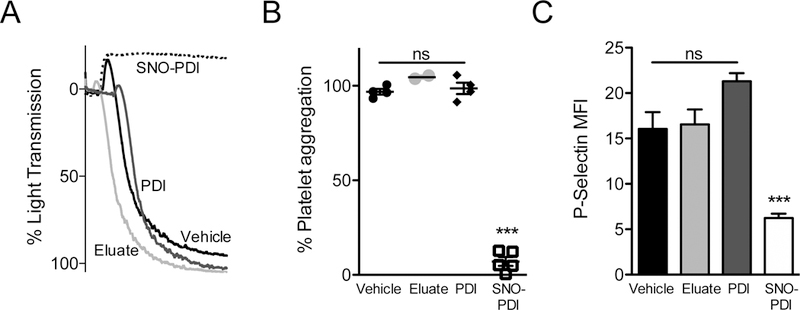
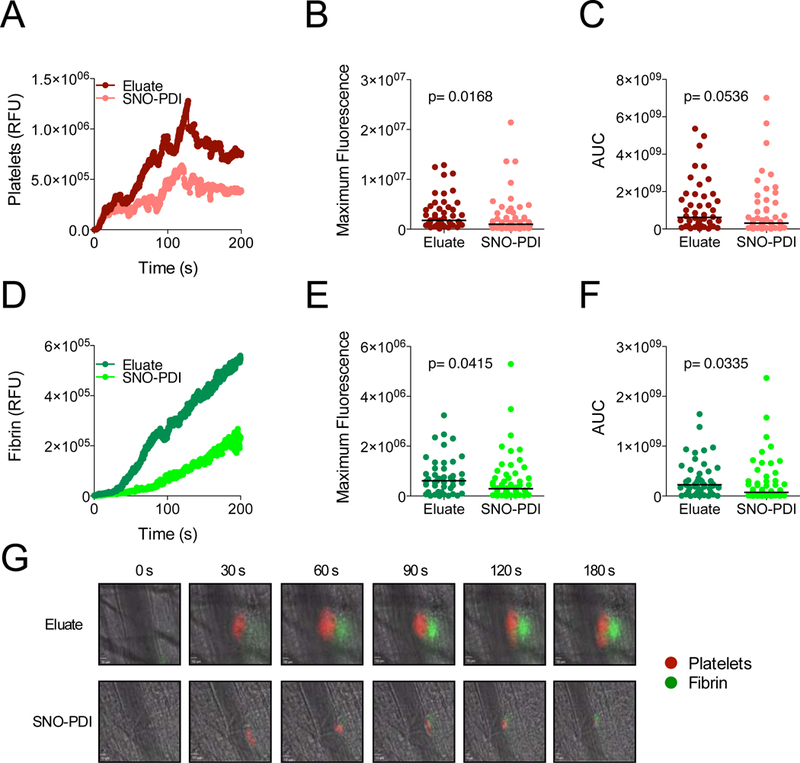
Essentials Nitric oxide synthesis controls protein disulfide isomerase (PDI) function. Nitric oxide (NO) modulation of PDI controls endothelial thrombogenicity. S-nitrosylated PDI inhibits platelet function and thrombosis. Nitric oxide maintains vascular quiescence in part through inhibition of PDI. SUMMARY: Background Protein disulfide isomerase (PDI) plays an essential role in thrombus formation, and PDI inhibition is being evaluated clinically as a novel anticoagulant strategy. However, little is known about the regulation of PDI in the vasculature. Thiols within the catalytic motif of PDI are essential for its role in thrombosis. These same thiols bind nitric oxide (NO), which is a potent regulator of vessel function. To determine whether regulation of PDI represents a mechanism by which NO controls vascular quiescence, we evaluated the effect of NO on PDI function in endothelial cells and platelets, and thrombus formation in vivo. Aim To assess the effect of S-nitrosylation on the regulation of PDI and other thiol isomerases in the vasculature. Methods and results The role of endogenous NO in PDI activity was evaluated by incubating endothelium with an NO scavenger, which resulted in exposure of free thiols, increased thiol isomerase activity, and enhanced thrombin generation on the cell membrane. Conversely, exposure of endothelium to NO carriers or elevation of endogenous NO levels by induction of NO synthesis resulted in S-nitrosylation of PDI and decreased surface thiol reductase activity. S-nitrosylation of platelet PDI inhibited its reductase activity, and S-nitrosylated PDI interfered with platelet aggregation, α-granule release, and thrombin generation on platelets. S-nitrosylated PDI also blocked laser-induced thrombus formation when infused into mice. S-nitrosylated ERp5 and ERp57 were found to have similar inhibitory activity. Conclusions These studies identify NO as a critical regulator of vascular PDI, and show that regulation of PDI function is an important mechanism by which NO maintains vascular quiescence.
一氧化氮合成控制蛋白二硫键异构酶(PDI)功能。一氧化氮(NO)对 PDI 的调节控制内皮的血栓形成。S-亚硝基化的 PDI 抑制血小板功能和血栓形成。一氧化氮通过抑制 PDI 部分维持血管静止。
蛋白二硫键异构酶(PDI)在血栓形成中起重要作用,PDI 抑制作用正在临床上作为一种新的抗凝策略进行评估。然而,对于 PDI 在血管中的调节知之甚少。PDI 催化基序中的巯基对于其在血栓形成中的作用是必不可少的。这些相同的巯基结合一氧化氮(NO),NO 是血管功能的有效调节剂。为了确定 NO 控制血管静止是否代表其调节 PDI 的机制,我们评估了 NO 对内皮细胞和血小板中 PDI 功能以及体内血栓形成的影响。
评估 S-亚硝基化对 PDI 和血管中其他巯基异构酶的调节作用。
通过用一氧化氮清除剂孵育内皮细胞来评估内源性 NO 在 PDI 活性中的作用,这导致暴露自由巯基、增加巯基异构酶活性和增强细胞膜上的凝血酶生成。相反,暴露于内皮细胞的 NO 载体或通过诱导 NO 合成升高内源性 NO 水平导致 PDI 的 S-亚硝基化和表面硫醇还原酶活性降低。血小板 PDI 的 S-亚硝基化抑制其还原酶活性,S-亚硝基化的 PDI 干扰血小板聚集、α-颗粒释放和血小板上的凝血酶生成。S-亚硝基化的 PDI 也阻止了注入小鼠时的激光诱导血栓形成。发现 S-亚硝基化的 ERp5 和 ERp57 具有相似的抑制活性。
这些研究将 NO 确定为血管 PDI 的关键调节剂,并表明 PDI 功能的调节是 NO 维持血管静止的重要机制。