Szécsényi Ágnes, Li Guanna, Gascon Jorge, Pidko Evgeny A
Catalysis Engineering Group, Chemical Engineering Department, and Inorganic Systems Engineering Group, Chemical Engineering Department, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands.
Catalysis Center, Advanced Catalytic Materials, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia.
ACS Catal. 2018 Sep 7;8(9):7961-7972. doi: 10.1021/acscatal.8b01672. Epub 2018 Jul 18.
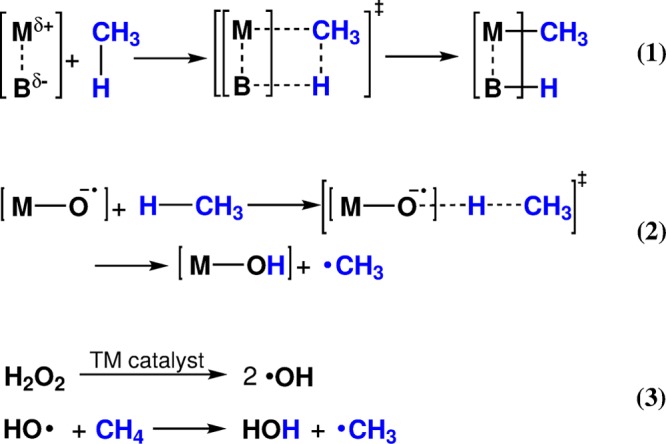
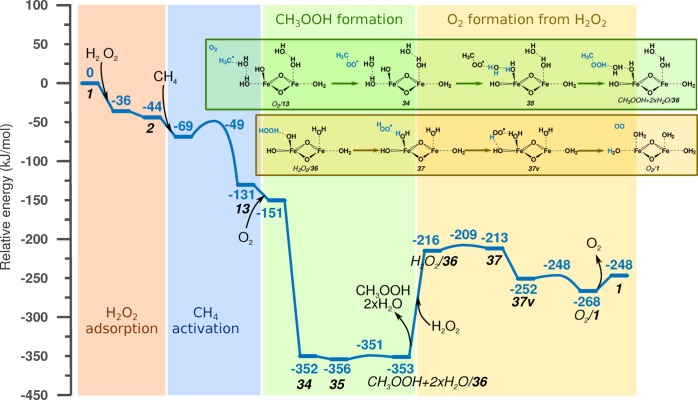
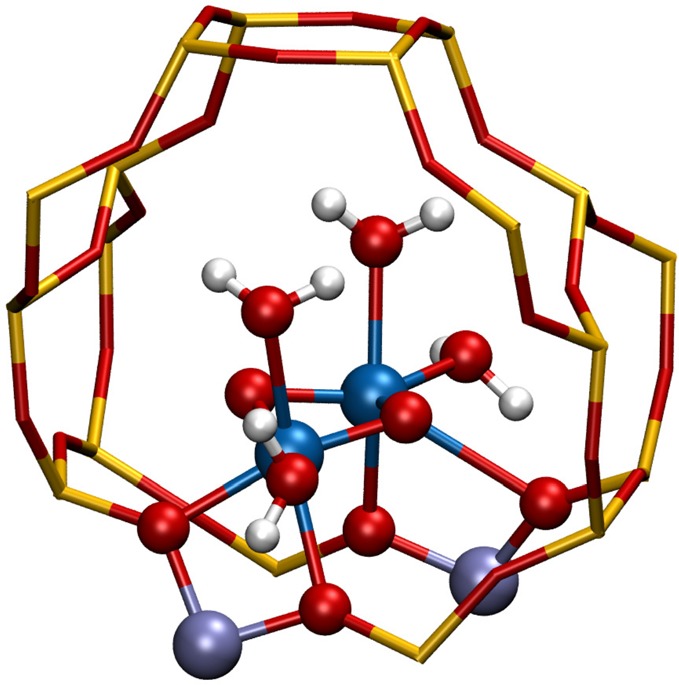
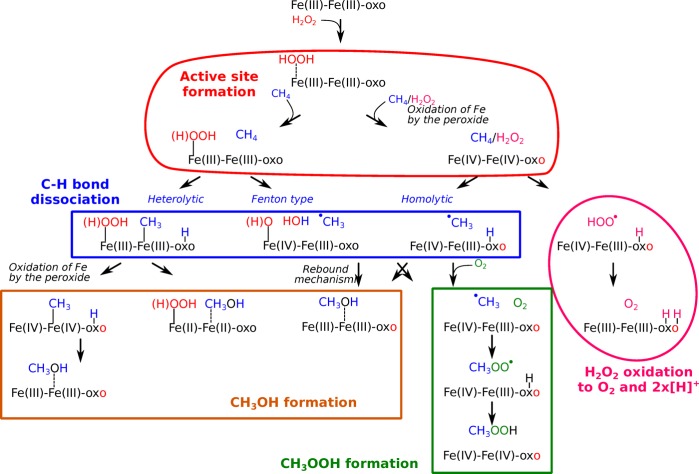
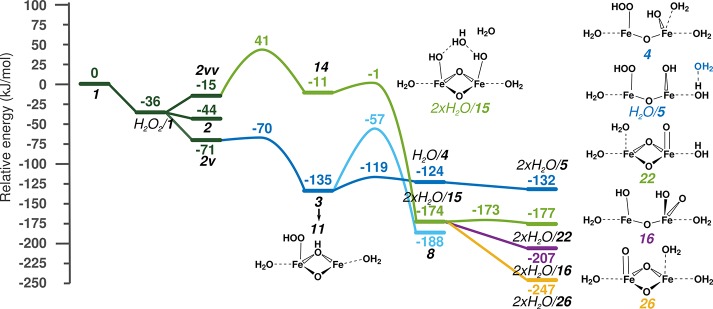
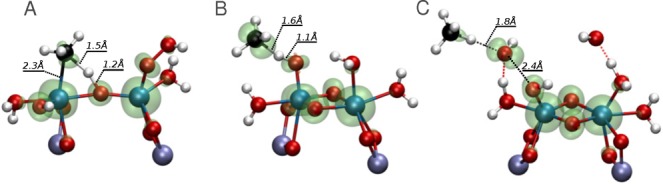
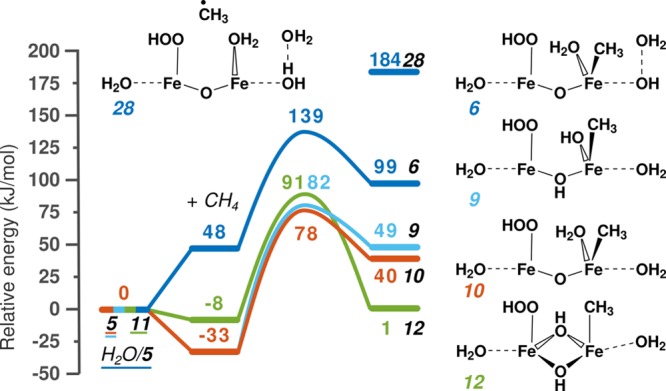
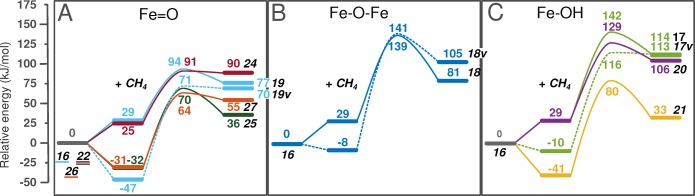
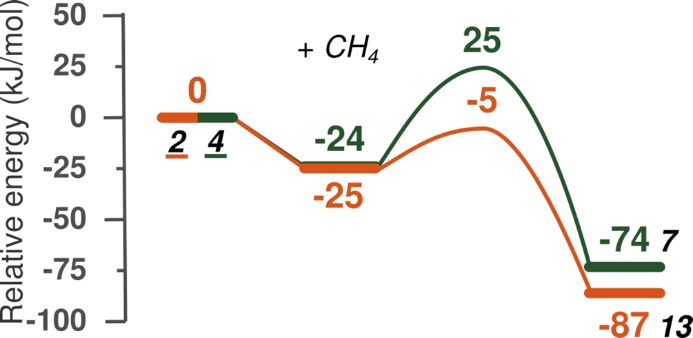
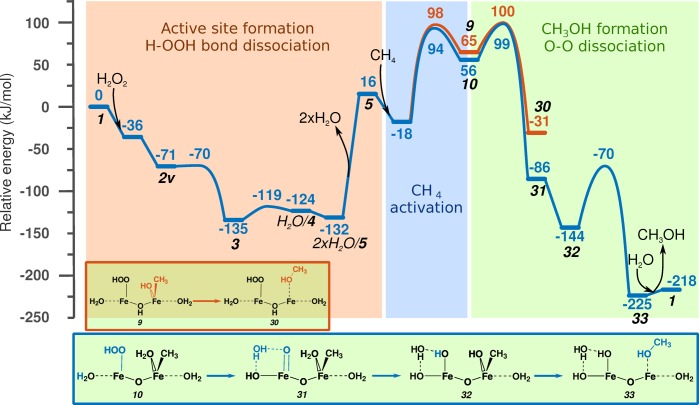
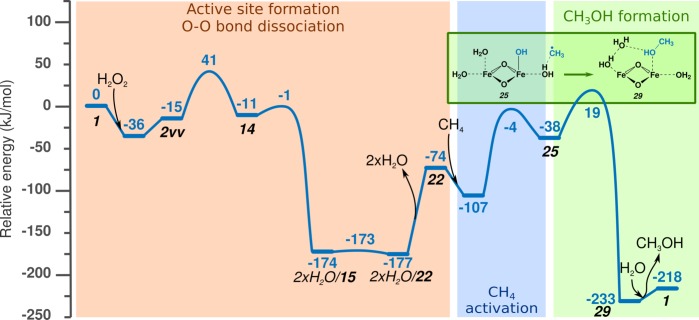
Periodic density functional theory (DFT) calculations were carried out to investigate the mechanism of methane oxidation with HO over the defined Fe sites in Fe/ZSM-5 zeolite. The initial Fe site is modeled as a [(HO)-Fe(III)-(μO)-Fe(III)-(HO)] extraframework cluster deposited in the zeolite pore and charge-compensated by two anionic lattice sites. The activation of this cluster with HO gives rise to the formation of a variety of Fe(III)-oxo and Fe(IV)-oxo complexes potentially reactive toward methane dissociation. These sites are all able to promote the first C-H bond cleavage in methane by following three possible reaction mechanisms: namely, (a) heterolytic and (b) homolytic methane dissociation as well as (c) Fenton-type reaction involving free OH radicals as the catalytic species. The C-H activation step is followed by formation of MeOH and MeOOH and regeneration of the active site. The Fenton-type path is found to proceed with the lowest activation barrier. Although the barriers for the alternative heterolytic and homolytic pathways are found to be somewhat higher, they are still quite favorable and are expected to be feasible under reaction conditions, resulting ultimately in MeOH and MeOOH products. HO oxidant competes with CH substrate for the same sites. Since the oxidation of HO to O and two [H] is energetically more favorable than the C-H oxofunctionalization, the overall efficiency of the latter target process remains low.
采用周期性密度泛函理论(DFT)计算,研究了在Fe/ZSM - 5沸石中特定Fe位点上HO与甲烷氧化反应的机理。初始Fe位点被模拟为沉积在沸石孔中的[(HO)-Fe(III)-(μO)-Fe(III)-(HO)]骨架外簇,并由两个阴离子晶格位点进行电荷补偿。该簇与HO的活化导致形成各种可能对甲烷解离具有反应活性的Fe(III)-氧和Fe(IV)-氧配合物。这些位点都能够通过以下三种可能的反应机制促进甲烷中第一个C - H键的裂解:即(a)异裂和(b)均裂甲烷解离以及(c)涉及游离OH自由基作为催化物种的芬顿型反应。C - H活化步骤之后是甲醇和甲氧基过氧化氢的形成以及活性位点的再生。发现芬顿型路径的活化能垒最低。尽管发现替代的异裂和均裂途径的能垒略高,但它们仍然相当有利,预计在反应条件下是可行的,最终生成甲醇和甲氧基过氧化氢产物。HO氧化剂与CH底物竞争相同的位点。由于HO氧化为O和两个[H]在能量上比C - H氧官能化更有利,因此后一目标过程的整体效率仍然较低。