Center for Molecular Medicine and Genetics, Wayne State University, Detroit, Michigan 48202, USA.
Department of Genetics, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA.
Genome Res. 2018 Nov;28(11):1701-1708. doi: 10.1101/gr.237354.118. Epub 2018 Sep 25.
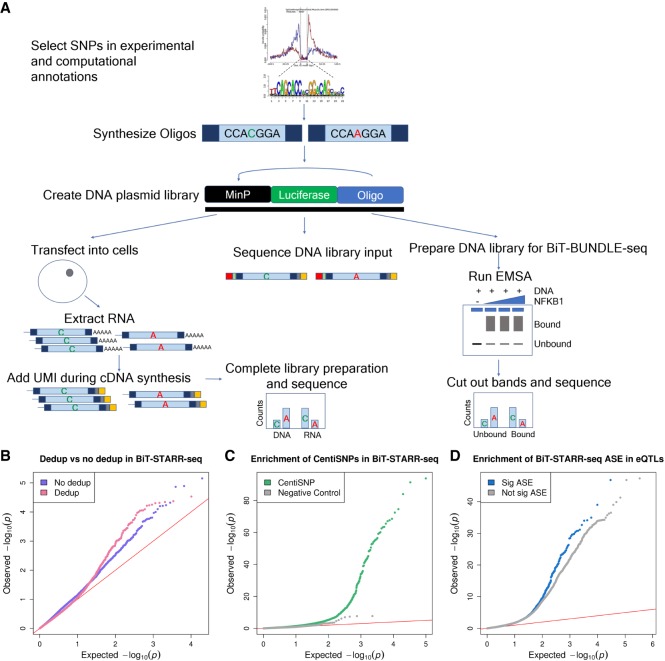
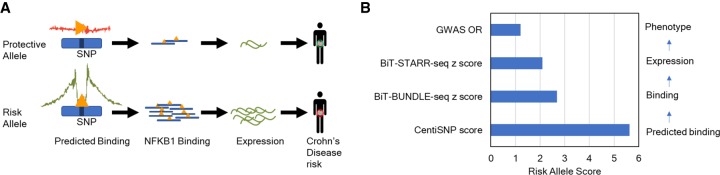
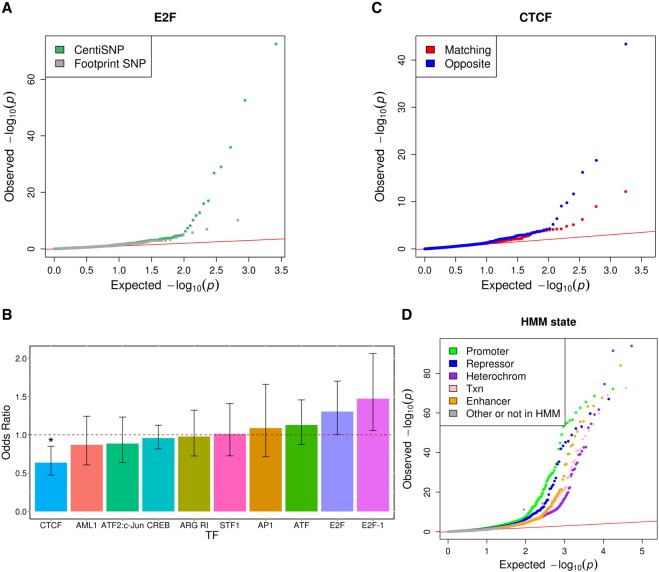
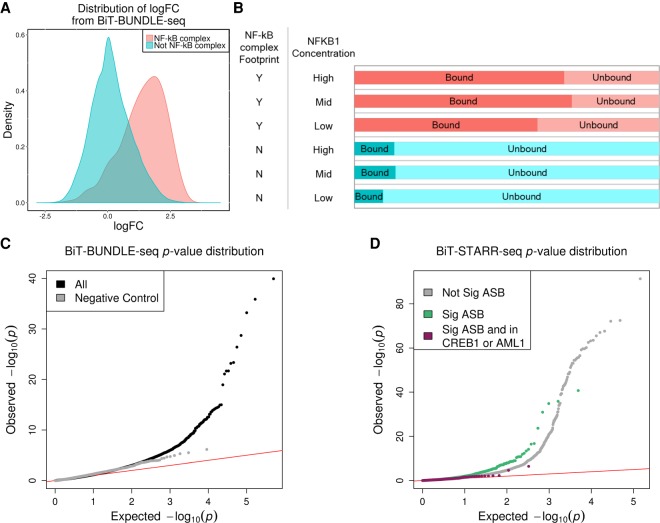
Many variants associated with complex traits are in noncoding regions and contribute to phenotypes by disrupting regulatory sequences. To characterize these variants, we developed a streamlined protocol for a high-throughput reporter assay, Biallelic Targeted STARR-seq (BiT-STARR-seq), that identifies allele-specific expression (ASE) while accounting for PCR duplicates through unique molecular identifiers. We tested 75,501 oligos (43,500 SNPs) and identified 2720 SNPs with significant ASE (FDR < 10%). To validate disruption of binding as one of the mechanisms underlying ASE, we developed a new high-throughput allele-specific binding assay for NFKB1. We identified 2684 SNPs with allele-specific binding (ASB) (FDR < 10%); 256 of these SNPs also had ASE (OR = 1.97, -value = 0.0006). Of variants associated with complex traits, 1531 resulted in ASE, and 1662 showed ASB. For example, we characterized that the Crohn's disease risk variant for rs3810936 increases NFKB1 binding and results in altered gene expression.
许多与复杂性状相关的变异位于非编码区域,通过破坏调控序列对表型产生影响。为了研究这些变异,我们开发了一种用于高通量报告基因检测的简化方案,即双等位基因靶向 STARR-seq(BiT-STARR-seq),该方案可通过独特的分子标识符识别等位基因特异性表达(ASE),同时考虑到 PCR 重复。我们测试了 75501 个寡核苷酸(43500 个 SNP),并鉴定出 2720 个具有显著 ASE(FDR<10%)的 SNP。为了验证结合破坏是 ASE 潜在机制之一,我们开发了一种新的 NFKB1 等位基因特异性结合的高通量检测方法。我们鉴定出 2684 个具有等位基因特异性结合(ASB)的 SNP(FDR<10%);其中 256 个 SNP 也具有 ASE(OR=1.97,-value=0.0006)。在与复杂性状相关的变异中,有 1531 个导致 ASE,1662 个表现出 ASB。例如,我们发现克罗恩病风险变异 rs3810936 增加了 NFKB1 结合,导致基因表达改变。