Pouya Farzaneh, Mojtabanezhad Shariatpanahi Afsaneh, Ghaffarzadegan Kamran, Tabatabaee Yazdi Seyed Abbas, Golmohammadzadeh Hamed, Soltani Ghodratollah, Aminian Toosi Kian, Kerachian Mohammad Amin
Department of Medical Genetics, Faculty of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran.
Cancer Genetics Research Unit, Reza Radiotherapy and Oncology Center, Mashhad, Iran.
Mol Genet Genomic Med. 2018 Nov;6(6):1031-1040. doi: 10.1002/mgg3.479. Epub 2018 Sep 26.
Familial adenomatous polyposis (FAP) is a familial colorectal cancer predisposition syndrome characterized by the development of numerous colorectal polyps, which is inherited in an autosomal dominant manner. FAP is caused by germ line mutations in adenomatous polyposis coli (APC) gene. Here, we described the identification of a causative APC gene deletion associated with FAP in an Iranian family.
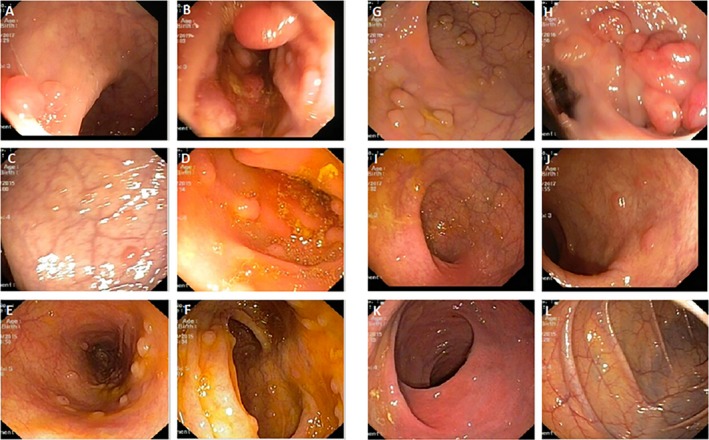
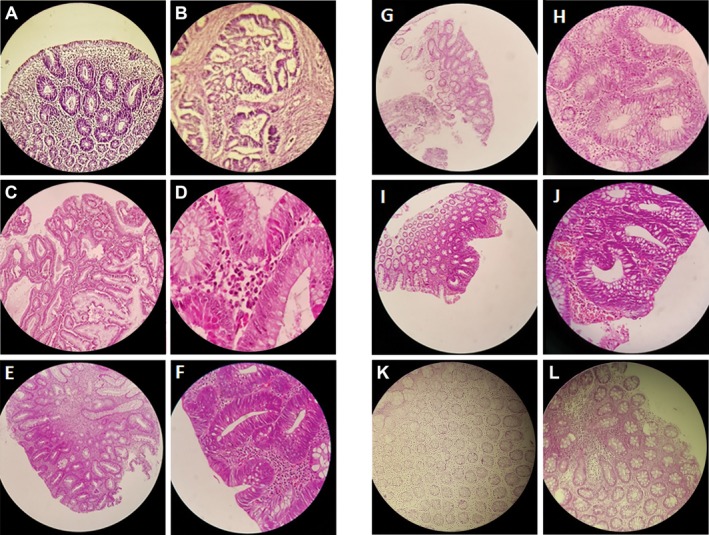
Diagnosis of FAP was based on clinical findings, family history, and medical records (colonoscopy and histopathological data) after the patients were referred to Reza Radiotherapy and Oncology Center, Iran, for colonoscopy. Blood samples were collected, and genomic DNA was extracted. APC mutation screening was conducted by target next-generation sequencing and quantitative real-time PCR.
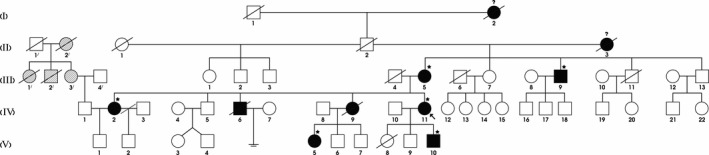
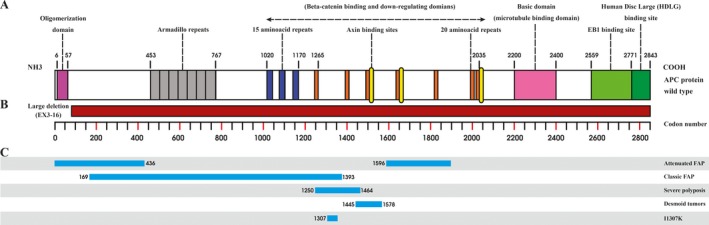
A novel heterozygous large deletion mutation, c.(135+1_136-1)(*2113+1*2114-1) spanning exon 3 to 16 [EX3_16 DEL] of APC gene (GenBank Accession# MG712911), was detected in a proband and all her affected relatives in five generations, which was absent in unaffected family members and normal controls.
This novel deletion is the first report, describing the largest deletion of APC gene. Our novel finding contributes to a more comprehensive database of germ line mutations of APC gene that could be used in medical practice for the molecular diagnosis, risk assessment susceptibility of the disease for the FAP patients.
家族性腺瘤性息肉病(FAP)是一种家族性结直肠癌易患综合征,其特征是出现大量结直肠息肉,以常染色体显性方式遗传。FAP由腺瘤性息肉病 coli(APC)基因的种系突变引起。在此,我们描述了在一个伊朗家庭中鉴定出与FAP相关的致病性APC基因缺失。
在患者被转诊至伊朗的雷扎放射治疗与肿瘤中心进行结肠镜检查后,根据临床发现、家族史和病历(结肠镜检查及组织病理学数据)对FAP进行诊断。采集血样并提取基因组DNA。通过靶向二代测序和定量实时PCR进行APC突变筛查。
在一名先证者及其五代内所有受影响的亲属中检测到一种新的杂合性大片段缺失突变,c.(135 + 1_136 - 1)(*2113 + 1*2114 - 1),跨越APC基因的外显子3至16 [EX3_16 DEL](GenBank登录号#MG712911),未受影响的家庭成员和正常对照中不存在该突变。
这种新的缺失是首次报道,描述了APC基因最大的缺失。我们的新发现有助于建立更全面的APC基因种系突变数据库,可用于医疗实践中对FAP患者进行分子诊断、疾病风险评估及易感性分析。