Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Department of Clinical Sciences and Community Health, University of Milan, Italy
Department of Haematology, University College London, UK.
Haematologica. 2019 Mar;104(3):477-484. doi: 10.3324/haematol.2018.198887. Epub 2018 Oct 18.
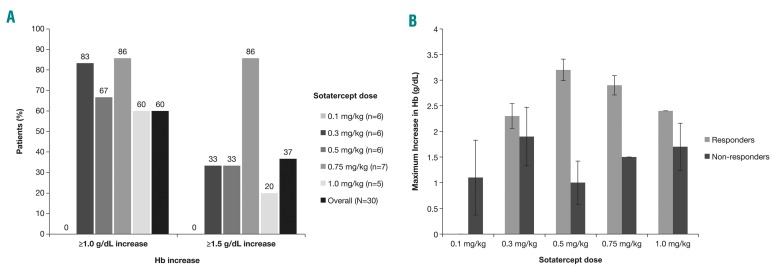
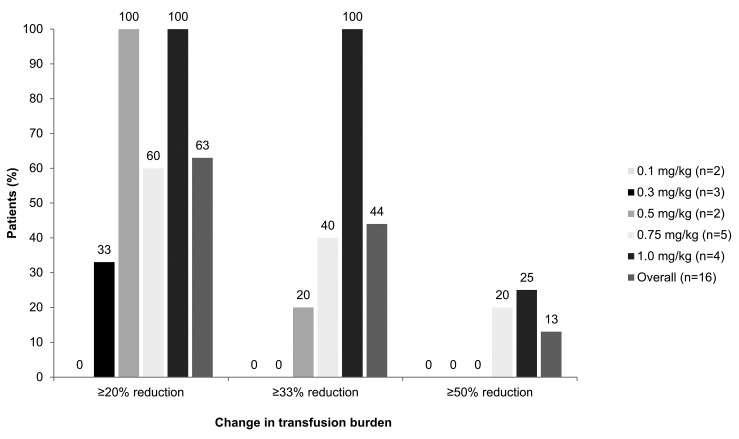
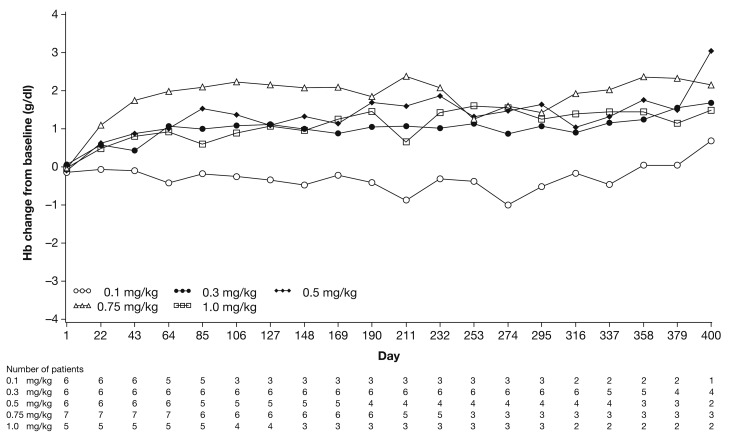
β-thalassemia, a hereditary blood disorder caused by defective synthesis of hemoglobin β globin chains, leads to ineffective erythropoiesis and chronic anemia that may require blood transfusions. Sotatercept (ACE-011) acts as a ligand trap to inhibit negative regulators of late-stage erythropoiesis in the transforming growth factor β superfamily, correcting ineffective erythropoiesis. In this phase II, open-label, dose-finding study, 16 patients with transfusion-dependent β -thalassemia and 30 patients with non-transfusion-dependent β-thalassemia were enrolled at seven centers in four countries between November 2012 and November 2014. Patients were treated with sotatercept at doses of 0.1, 0.3, 0.5, 0.75, or 1.0 mg/kg to determine a safe and effective dose. Doses were administered by subcutaneous injection every 3 weeks. Patients were treated for ≤22 months. Response was assessed as a ≥20% reduction in transfusion burden sustained for 24 weeks in transfusion-dependent β-thalassemia patients, and an increase in hemoglobin level of ≥1.0 g/dL sustained for 12 weeks in non-transfusion-dependent β-thalassemia patients. Sotatercept was well tolerated. After a median treatment duration of 14.4 months (range 0.6-35.9), no severe life-threatening adverse events were observed. Thirteen percent of patients reported serious but manageable adverse events. The active dose of sotatercept was ≥0.3 mg/kg for patients with non-transfusion-dependent β-thalassemia and ≥0.5 mg/kg for those with transfusion-dependent β-thalassemia. Of 30 non-transfusion-dependent β-thalassemia patients treated with ≥0.1 mg/kg sotatercept, 18 (60%) achieved a mean hemoglobin increase ≥1.0 g/dL, and 11 (37%) an increase ≥1.5 g/dL, sustained for ≥12 weeks. Four (100%) transfusion-dependent β-thalassemia patients treated with 1.0 mg/kg sotatercept achieved a transfusion-burden reduction of ≥20%. Sotatercept was effective and well tolerated in patients with β-thalassemia. Most patients with non-transfusion-dependent β-thalassemia treated with higher doses achieved sustained increases in hemoglobin level. Transfusion-dependent β-thalassemia patients treated with higher doses of sotatercept achieved notable reductions in transfusion requirements. This trial was registered at ClinicalTrials.gov with the number NCT01571635.
β-地中海贫血是一种遗传性血液疾病,由血红蛋白β珠蛋白链的缺陷合成引起,导致无效的红细胞生成和慢性贫血,可能需要输血。Sotatercept(ACE-011)作为一种配体陷阱,可抑制转化生长因子β超家族中晚期红细胞生成的负调节剂,纠正无效的红细胞生成。在这项 2 期、开放标签、剂量发现研究中,16 名依赖输血的β地中海贫血患者和 30 名非输血依赖的β地中海贫血患者在 2012 年 11 月至 2014 年 11 月期间在四个国家的七个中心入组。患者接受 sotatercept 治疗,剂量为 0.1、0.3、0.5、0.75 或 1.0 mg/kg,以确定安全有效的剂量。每周通过皮下注射给药 3 次。患者治疗≤22 个月。在依赖输血的β地中海贫血患者中,评估反应为持续 24 周的输血负担减少≥20%,非输血依赖的β地中海贫血患者中血红蛋白水平增加≥1.0 g/dL 持续 12 周。Sotatercept 耐受性良好。在中位治疗时间 14.4 个月(范围 0.6-35.9)后,未观察到严重危及生命的不良事件。13%的患者报告了严重但可管理的不良事件。对于非输血依赖的β地中海贫血患者,活性剂量≥0.3 mg/kg,对于依赖输血的β地中海贫血患者,活性剂量≥0.5 mg/kg。在接受≥0.1 mg/kg sotatercept 治疗的 30 名非输血依赖的β地中海贫血患者中,18 名(60%)实现了平均血红蛋白增加≥1.0 g/dL,11 名(37%)增加≥1.5 g/dL,持续≥12 周。接受 1.0 mg/kg sotatercept 治疗的 4 名(100%)依赖输血的β地中海贫血患者实现了输血负担减少≥20%。Sotatercept 在β地中海贫血患者中有效且耐受良好。大多数接受高剂量治疗的非输血依赖的β地中海贫血患者实现了血红蛋白水平的持续升高。接受 sotatercept 高剂量治疗的依赖输血的β地中海贫血患者显著减少了输血需求。该试验在 ClinicalTrials.gov 注册,编号为 NCT01571635。