Department of Statistics, Stanford University, Stanford, 94305, CA, USA.
Department of Statistics, The University of Chicago, Chicago, 60637, IL, USA.
Nat Commun. 2018 Oct 19;9(1):4361. doi: 10.1038/s41467-018-06805-x.
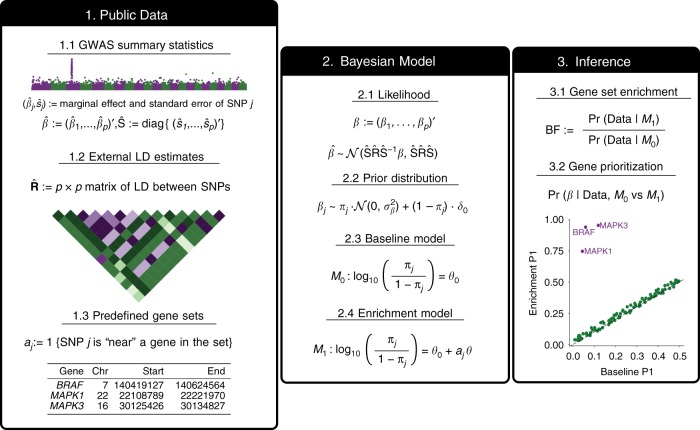
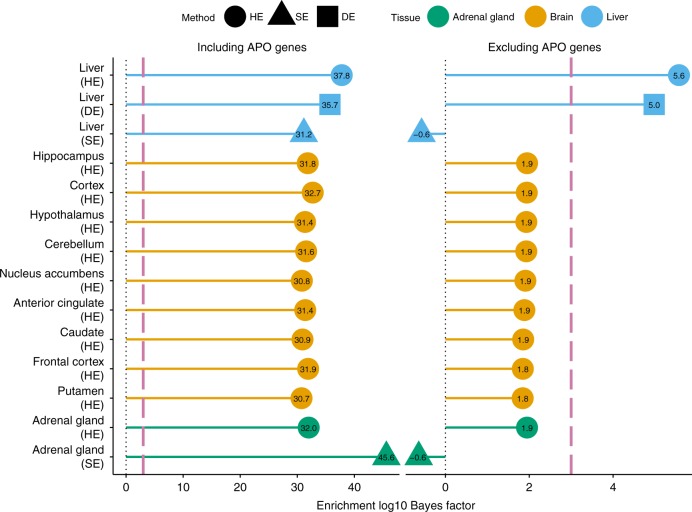
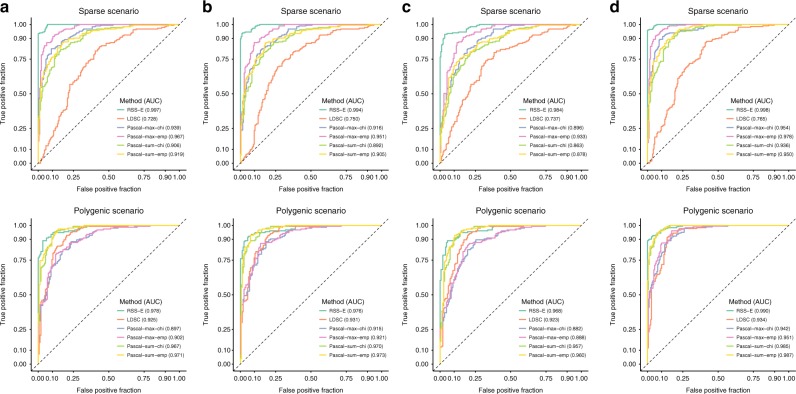
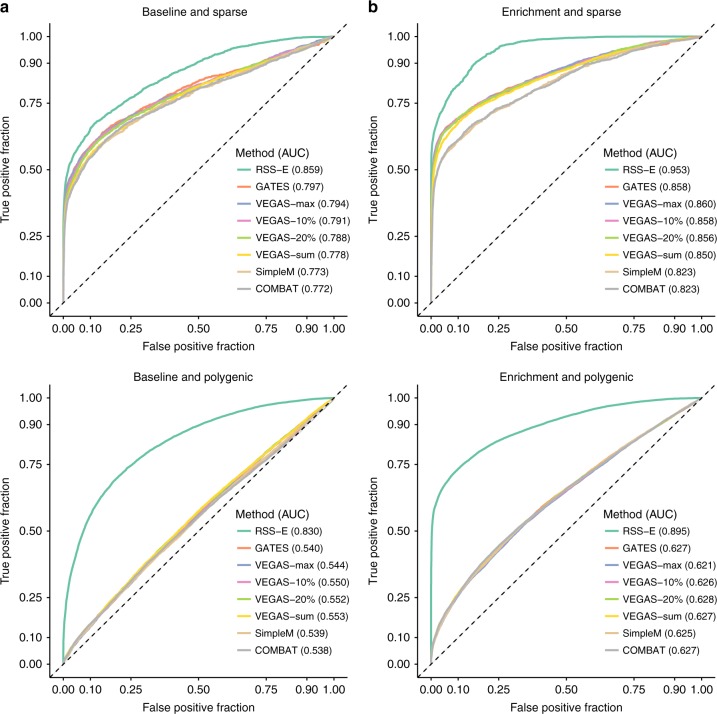
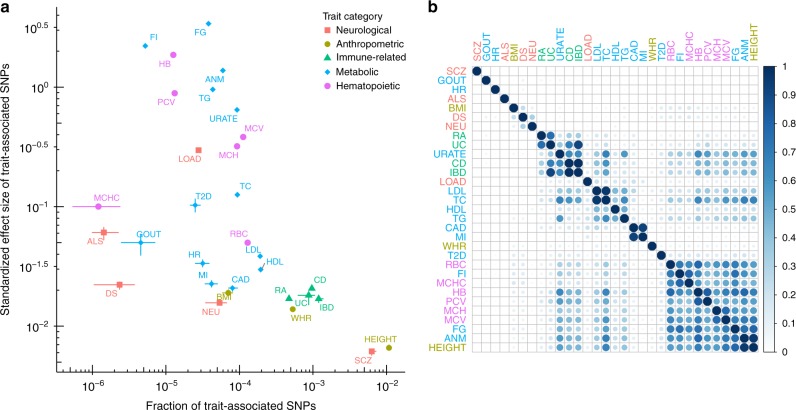
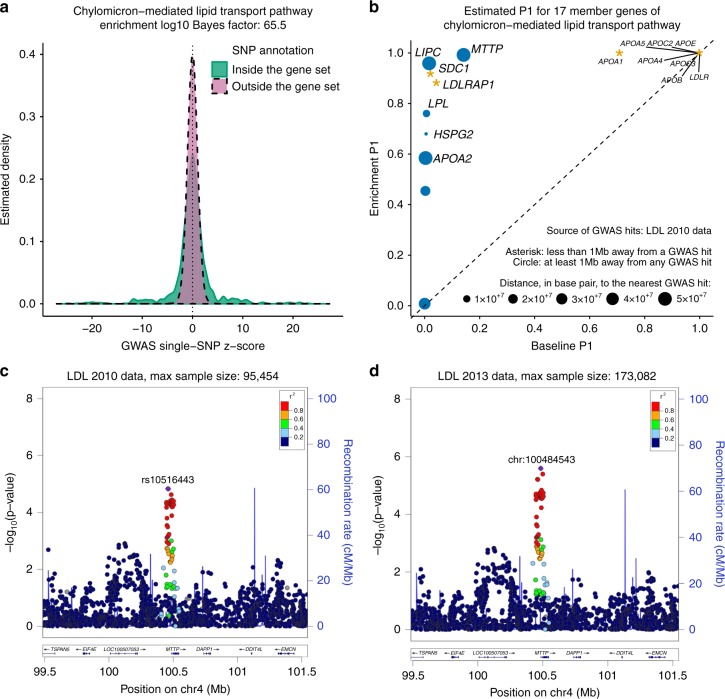
Genome-wide association studies (GWAS) aim to identify genetic factors associated with phenotypes. Standard analyses test variants for associations individually. However, variant-level associations are hard to identify and can be difficult to interpret biologically. Enrichment analyses help address both problems by targeting sets of biologically related variants. Here we introduce a new model-based enrichment method that requires only GWAS summary statistics. Applying this method to interrogate 4,026 gene sets in 31 human phenotypes identifies many previously-unreported enrichments, including enrichments of endochondral ossification pathway for height, NFAT-dependent transcription pathway for rheumatoid arthritis, brain-related genes for coronary artery disease, and liver-related genes for Alzheimer's disease. A key feature of our method is that inferred enrichments automatically help identify new trait-associated genes. For example, accounting for enrichment in lipid transport genes highlights association between MTTP and low-density lipoprotein levels, whereas conventional analyses of the same data found no significant variants near this gene.
全基因组关联研究(GWAS)旨在识别与表型相关的遗传因素。标准分析逐个测试变体的关联。然而,变体水平的关联很难识别,并且在生物学上也很难解释。通过针对具有生物学相关性的变体集,富集分析有助于解决这两个问题。在这里,我们介绍了一种新的基于模型的富集方法,该方法仅需要 GWAS 汇总统计信息。将此方法应用于 31 个人类表型中的 4026 个基因集的探究,确定了许多以前未报道过的富集,包括身高的软骨内骨化途径、类风湿关节炎的 NFAT 依赖性转录途径、冠心病的大脑相关基因和阿尔茨海默病的肝脏相关基因。我们的方法的一个关键特征是,推断出的富集会自动帮助识别新的与性状相关的基因。例如,脂质转运基因的富集突出了 MTTP 与低密度脂蛋白水平之间的关联,而对相同数据的常规分析并未发现该基因附近存在显着变体。