Bhatia Vaibhav, Valdés-Sánchez Lourdes, Rodriguez-Martinez Daniel, Bhattacharya Shom Shankar
CABIMER (Centro Andaluz de Biología Molecular y Medicina Regenerativa), (FPS) Fundacion Progreso y Salud, Sevilla, Andalucia, 41092, Spain.
F1000Res. 2018 Aug 10;7:1233. doi: 10.12688/f1000research.15579.1. eCollection 2018.
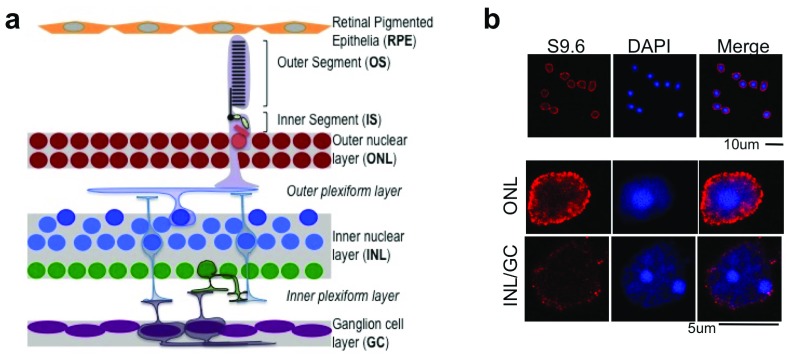
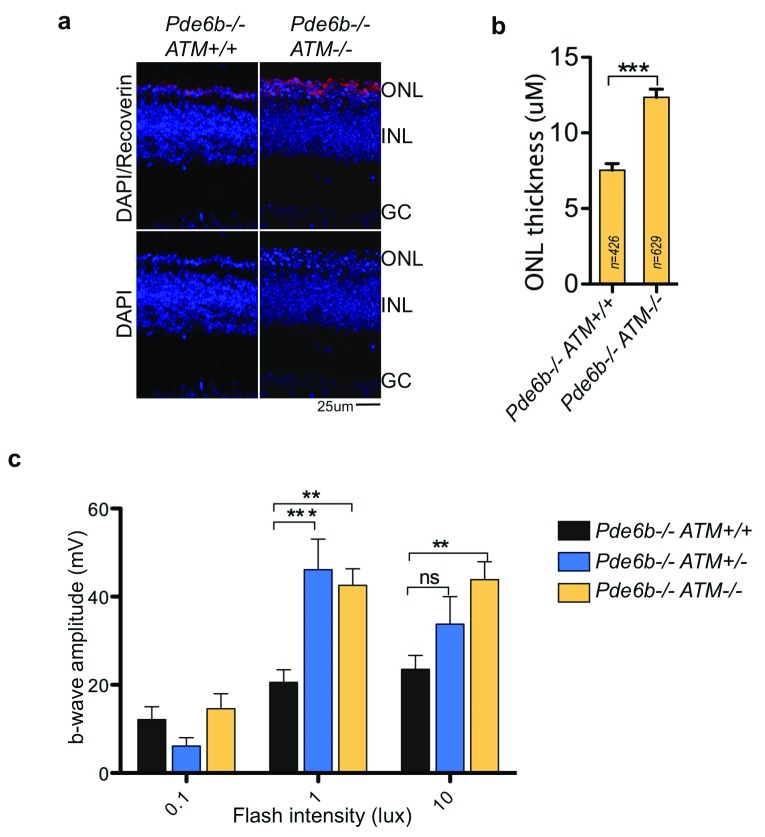
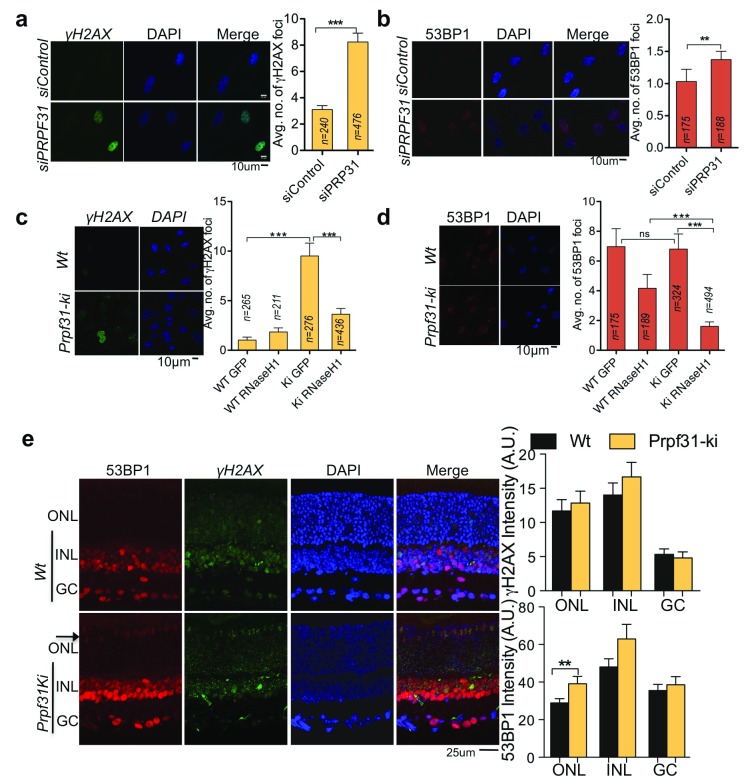
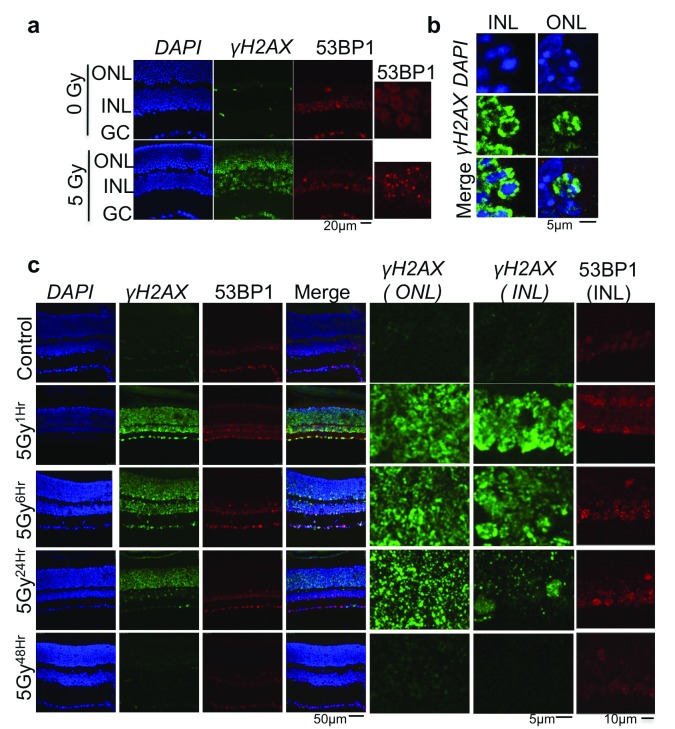
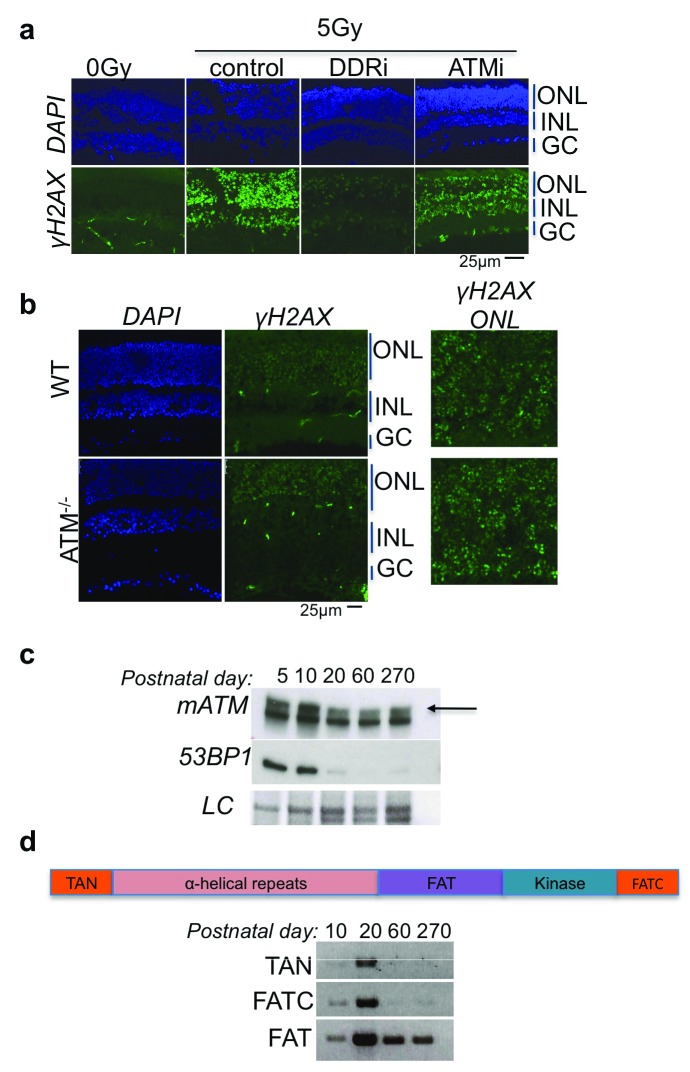
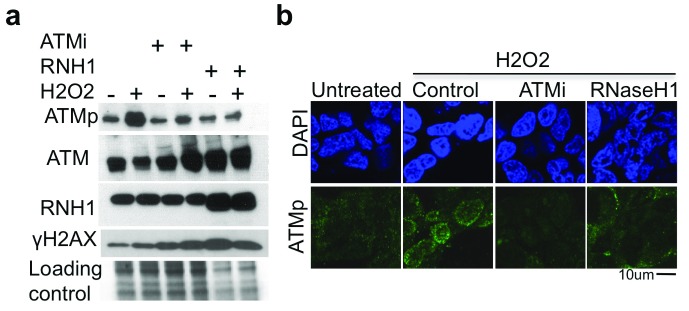
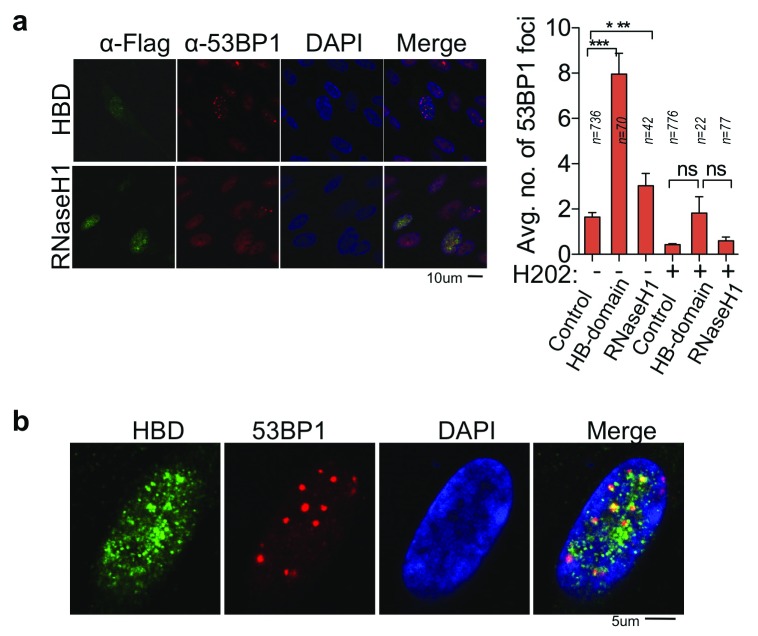
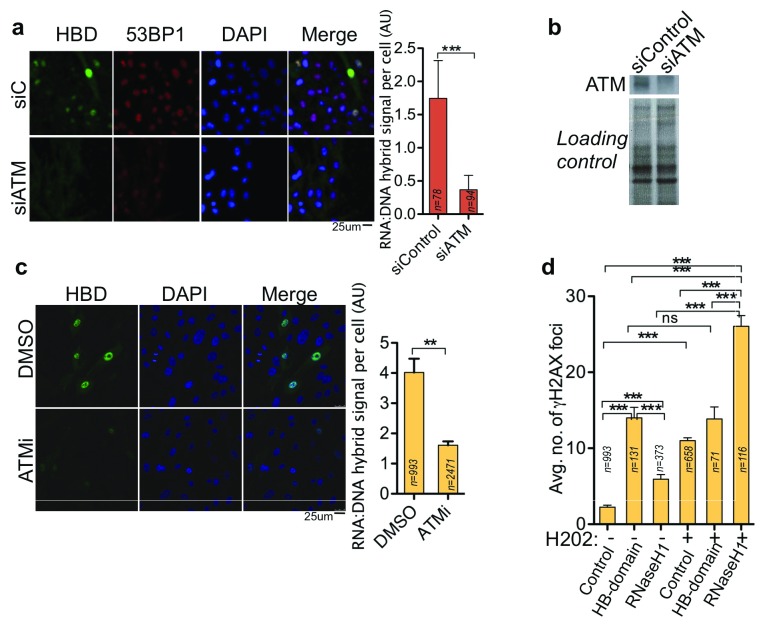
Photoreceptors, light-sensing neurons in retina, are central to vision. Photoreceptor cell death (PCD) is observed in most inherited and acquired retinal dystrophies. But the underlying molecular mechanism of PCD is unclear. Photoreceptors are sturdy neurons that survive high oxidative and phototoxic stress, which are known threats to genome stability. Unexpectedly, DNA damage response in mice photoreceptors is compromised; mainly due to loss of crucial DNA repair proteins, ATM and 53BP1. We tried to understand the molecular function of ATM and 53BP1 in response to oxidative stress and how suppression of DNA repair response in mice retina affect photoreceptor cell survival. We use the state of art cell biology methods and structure-function analysis of mice retina. RNA:DNA hybrids (S9.6 antibody and Hybrid-binding domain of RNaseH1) and DNA repair foci (gH2AX and 53BP1) are quantified by confocal microscopy, in retinal sections and cultured cell lines. Oxidative stress, DNA double strand break, RNaseH1 expression and small-molecule kinase-inhibitors were used to understand the role of ATM and RNA:DNA hybrids in DNA repair. Lastly, retinal structure and function of ATM deficient mice, in Retinal degeneration 1 (Pde6brd1) background, is studied using Immunohistochemistry and Electroretinography. Our work has three novel findings: firstly, both human and mice photoreceptor cells specifically accumulate RNA:DNA hybrids, a structure formed by re-hybridization of nascent RNA with template DNA during transcription. Secondly, RNA:DNA-hybrids promote ataxia-telangiectasia mutated (ATM) activation during oxidative stress and 53BP1-foci formation during downstream DNA repair process. Thirdly, loss of ATM -in murine photoreceptors- protract DNA repair but also promote their survival. We propose that due to high oxidative stress and accumulation of RNA:DNA-hybrids in photoreceptors, expression of ATM is tightly regulated to prevent PCD. Inefficient regulation of ATM expression could be central to PCD and inhibition of ATM-activation could suppress PCD in retinal dystrophy patients.
光感受器是视网膜中的光感神经元,是视觉的核心。在大多数遗传性和获得性视网膜营养不良中都观察到光感受器细胞死亡(PCD)。但PCD的潜在分子机制尚不清楚。光感受器是坚固的神经元,能够在高氧化和光毒性应激下存活,而这些应激是已知的对基因组稳定性的威胁。出乎意料的是,小鼠光感受器中的DNA损伤反应受损;主要是由于关键的DNA修复蛋白ATM和53BP1的缺失。我们试图了解ATM和53BP1在应对氧化应激时的分子功能,以及小鼠视网膜中DNA修复反应的抑制如何影响光感受器细胞的存活。我们使用先进的细胞生物学方法和对小鼠视网膜进行结构-功能分析。通过共聚焦显微镜对视网膜切片和培养细胞系中的RNA:DNA杂交体(S9.6抗体和RNaseH1的杂交结合结构域)和DNA修复灶(gH2AX和53BP1)进行定量。利用氧化应激、DNA双链断裂、RNaseH1表达和小分子激酶抑制剂来了解ATM和RNA:DNA杂交体在DNA修复中的作用。最后,使用免疫组织化学和视网膜电图研究了视网膜变性1(Pde6brd1)背景下ATM缺陷小鼠的视网膜结构和功能。我们的研究有三个新发现:首先,人类和小鼠的光感受器细胞都特异性地积累RNA:DNA杂交体,这是一种在转录过程中新生RNA与模板DNA重新杂交形成的结构。其次,RNA:DNA杂交体在氧化应激期间促进共济失调毛细血管扩张突变(ATM)的激活,并在下游DNA修复过程中促进53BP1灶的形成。第三,小鼠光感受器中ATM的缺失会延长DNA修复,但也能促进它们的存活。我们提出,由于光感受器中高氧化应激和RNA:DNA杂交体的积累,ATM的表达受到严格调控以防止PCD。ATM表达的低效调控可能是PCD的核心,而抑制ATM激活可能会抑制视网膜营养不良患者的PCD。