Meningitis and Vaccine Preventable Diseases Branch, Centers for Disease Control and Prevention, Atlanta, Georgia, USA.
Division of Infectious Diseases, Department of Medicine, Emory University School of Medicine, Atlanta, Georgia, USA.
Sci Rep. 2018 Oct 25;8(1):15803. doi: 10.1038/s41598-018-33622-5.
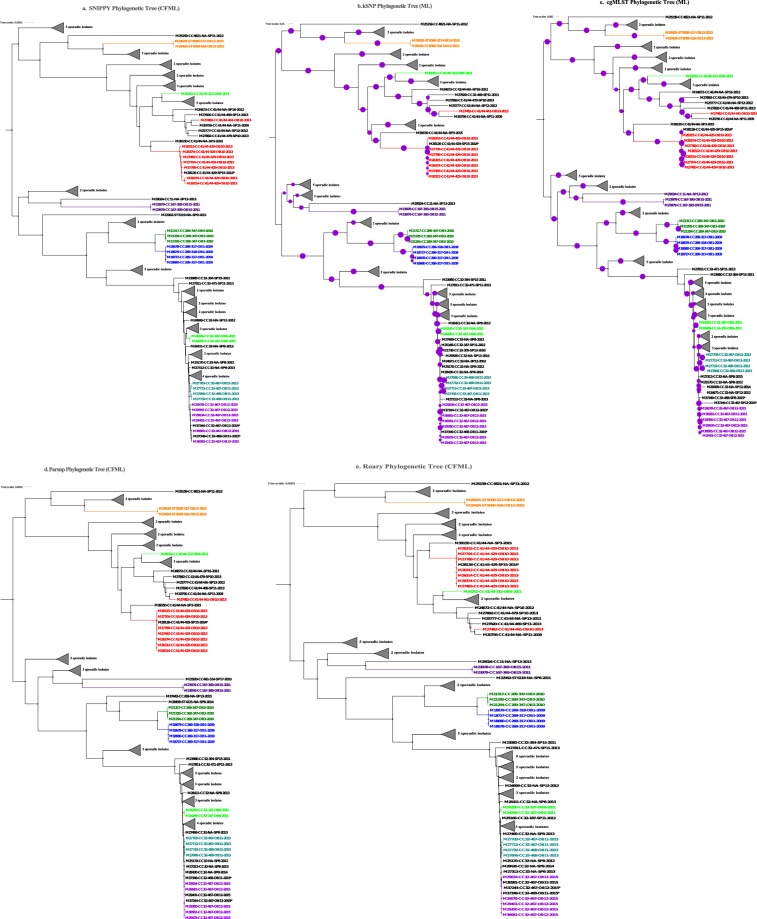
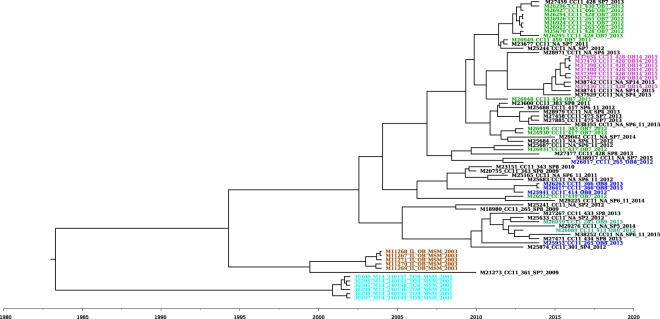
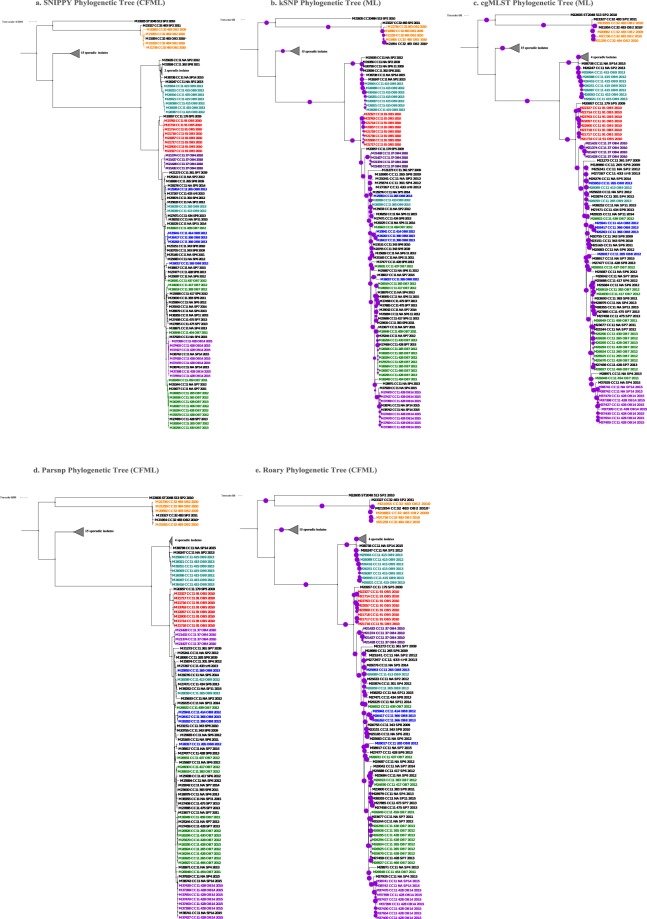
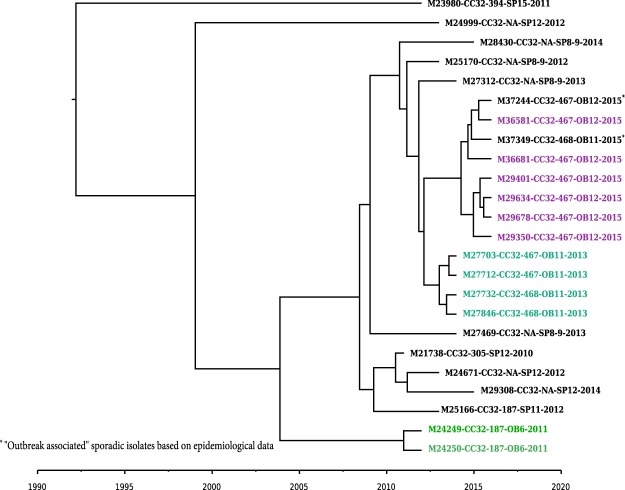
Although rare in the U.S., outbreaks due to Neisseria meningitidis do occur. Rapid, early outbreak detection is important for timely public health response. In this study, we characterized U.S. meningococcal isolates (N = 201) from 15 epidemiologically defined outbreaks (2009-2015) along with temporally and geographically matched sporadic isolates using multilocus sequence typing, pulsed-field gel electrophoresis (PFGE), and six whole genome sequencing (WGS) based methods. Recombination-corrected maximum likelihood (ML) and Bayesian phylogenies were reconstructed to identify genetically related outbreak isolates. All WGS analysis methods showed high degree of agreement and distinguished isolates with similar or indistinguishable PFGE patterns, or the same strain genotype. Ten outbreaks were caused by a single strain; 5 were due to multiple strains. Five sporadic isolates were phylogenetically related to 2 outbreaks. Analysis of 9 outbreaks using timed phylogenies identified the possible origin and estimated the approximate time that the most recent common ancestor emerged for outbreaks analyzed. U.S. meningococcal outbreaks were caused by single- or multiple-strain introduction, with organizational outbreaks mainly caused by a clonal strain and community outbreaks by divergent strains. WGS can infer linkage of meningococcal cases when epidemiological links are uncertain. Accurate identification of outbreak-associated cases requires both WGS typing and epidemiological data.
虽然在美国罕见,但脑膜炎奈瑟菌引起的暴发确实会发生。快速、早期的暴发检测对于及时的公共卫生应对非常重要。在这项研究中,我们使用多位点序列分型、脉冲场凝胶电泳(PFGE)和基于 6 种全基因组测序(WGS)的方法,对 15 起具有明确流行病学定义的暴发(2009-2015 年)中的 201 株美国脑膜炎奈瑟菌分离株以及在时间和地理上匹配的散发性分离株进行了特征描述。通过重组校正最大似然(ML)和贝叶斯系统发育树来识别具有遗传相关性的暴发分离株。所有 WGS 分析方法均高度一致,并区分了 PFGE 模式相似或无法区分的分离株,或相同的菌株基因型。10 起暴发由单一致病菌株引起;5 起由多株引起。5 株散发性分离株与 2 起暴发有关。使用时间系统发育树分析 9 起暴发,确定了可能的起源,并估计了分析的暴发最近共同祖先出现的大致时间。美国脑膜炎奈瑟菌暴发是由单一致病菌株或多株菌引起的,组织暴发主要由克隆菌株引起,社区暴发由不同菌株引起。WGS 可以在流行病学联系不确定时推断脑膜炎奈瑟菌病例的关联性。准确识别暴发相关病例需要 WGS 分型和流行病学数据。