Pettengill James B, Pightling Arthur W, Baugher Joseph D, Rand Hugh, Strain Errol
Biostatistics and Bioinformatics Staff, Center for Food Safety and Applied Nutrition, Food and Drug Administration, 5001 Campus Drive, College Park, MD 20740, United States of America.
PLoS One. 2016 Nov 10;11(11):e0166162. doi: 10.1371/journal.pone.0166162. eCollection 2016.
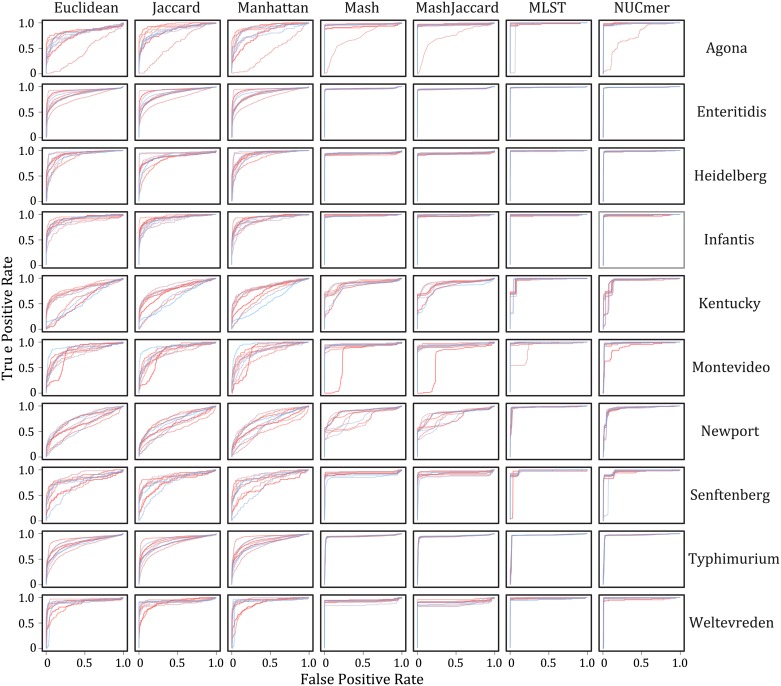
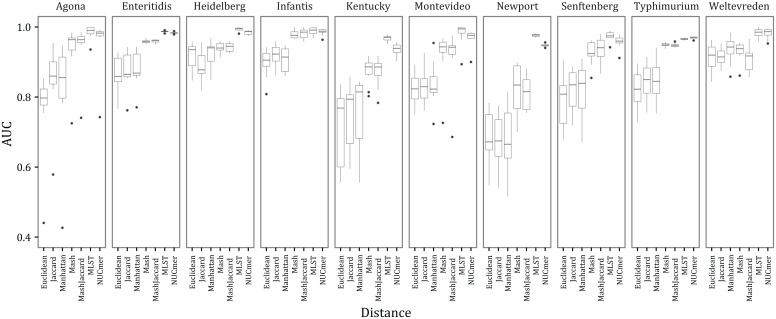
The adoption of whole-genome sequencing within the public health realm for molecular characterization of bacterial pathogens has been followed by an increased emphasis on real-time detection of emerging outbreaks (e.g., food-borne Salmonellosis). In turn, large databases of whole-genome sequence data are being populated. These databases currently contain tens of thousands of samples and are expected to grow to hundreds of thousands within a few years. For these databases to be of optimal use one must be able to quickly interrogate them to accurately determine the genetic distances among a set of samples. Being able to do so is challenging due to both biological (evolutionary diverse samples) and computational (petabytes of sequence data) issues. We evaluated seven measures of genetic distance, which were estimated from either k-mer profiles (Jaccard, Euclidean, Manhattan, Mash Jaccard, and Mash distances) or nucleotide sites (NUCmer and an extended multi-locus sequence typing (MLST) scheme). When analyzing empirical data (whole-genome sequence data from 18,997 Salmonella isolates) there are features (e.g., genomic, assembly, and contamination) that cause distances inferred from k-mer profiles, which treat absent data as informative, to fail to accurately capture the distance between samples when compared to distances inferred from differences in nucleotide sites. Thus, site-based distances, like NUCmer and extended MLST, are superior in performance, but accessing the computing resources necessary to perform them may be challenging when analyzing large databases.
在公共卫生领域采用全基因组测序对细菌病原体进行分子特征分析之后,人们越来越重视对新出现的疫情(如食源性沙门氏菌病)进行实时检测。相应地,全基因组序列数据的大型数据库正在不断充实。这些数据库目前包含数万个样本,预计在几年内将增长到数十万。为了使这些数据库得到最佳利用,必须能够快速查询它们,以准确确定一组样本之间的遗传距离。由于生物学(进化多样的样本)和计算(数PB的序列数据)问题,能够做到这一点具有挑战性。我们评估了七种遗传距离度量方法,这些方法是根据k-mer图谱(杰卡德距离、欧几里得距离、曼哈顿距离、Mash杰卡德距离和Mash距离)或核苷酸位点(NUCmer和扩展的多位点序列分型(MLST)方案)估算出来的。在分析经验数据(来自18997株沙门氏菌分离株的全基因组序列数据)时,存在一些特征(如基因组、组装和污染),这些特征导致从k-mer图谱推断出的距离(将缺失数据视为有信息的)在与从核苷酸位点差异推断出的距离相比时,无法准确捕捉样本之间的距离。因此,基于位点的距离,如NUCmer和扩展的MLST,在性能上更优,但在分析大型数据库时,获取执行这些方法所需的计算资源可能具有挑战性。