Theunissen Tom E J, Nguyen Minh, Kamps Rick, Hendrickx Alexandra T, Sallevelt Suzanne C E H, Gottschalk Ralph W H, Calis Chantal M, Stassen Alphons P M, de Koning Bart, Mulder-Den Hartog Elvira N M, Schoonderwoerd Kees, Fuchs Sabine A, Hilhorst-Hofstee Yvonne, de Visser Marianne, Vanoevelen Jo, Szklarczyk Radek, Gerards Mike, de Coo Irenaeus F M, Hellebrekers Debby M E I, Smeets Hubert J M
Department of Genetics and Cell Biology, Maastricht University Medical Centre, Maastricht, Netherlands.
Research Institute GROW, Maastricht University Medical Centre, Maastricht, Netherlands.
Front Genet. 2018 Oct 12;9:400. doi: 10.3389/fgene.2018.00400. eCollection 2018.
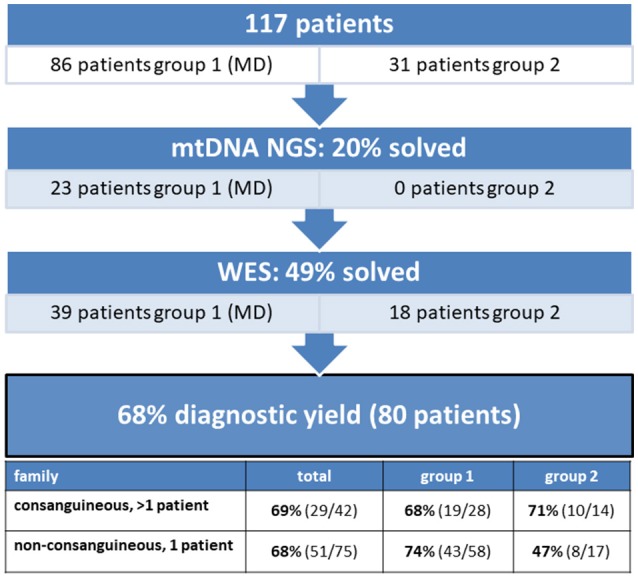
Mitochondrial disorders, characterized by clinical symptoms and/or OXPHOS deficiencies, are caused by pathogenic variants in mitochondrial genes. However, pathogenic variants in some of these genes can lead to clinical manifestations which overlap with other neuromuscular diseases, which can be caused by pathogenic variants in non-mitochondrial genes as well. Mitochondrial pathogenic variants can be found in the mitochondrial DNA (mtDNA) or in any of the 1,500 nuclear genes with a mitochondrial function. We have performed a two-step next-generation sequencing approach in a cohort of 117 patients, mostly children, in whom a mitochondrial disease-cause could likely or possibly explain the phenotype. A total of 86 patients had a mitochondrial disorder, according to established clinical and biochemical criteria. The other 31 patients had neuromuscular symptoms, where in a minority a mitochondrial genetic cause is present, but a non-mitochondrial genetic cause is more likely. All patients were screened for pathogenic variants in the mtDNA and, if excluded, analyzed by whole exome sequencing (WES). Variants were filtered for being pathogenic and compatible with an autosomal or X-linked recessive mode of inheritance in families with multiple affected siblings and/or consanguineous parents. Non-consanguineous families with a single patient were additionally screened for autosomal and X-linked dominant mutations in a predefined gene-set. We identified causative pathogenic variants in the mtDNA in 20% of the patient-cohort, and in nuclear genes in 49%, implying an overall yield of 68%. We identified pathogenic variants in mitochondrial and non-mitochondrial genes in both groups with, obviously, a higher number of mitochondrial genes affected in mitochondrial disease patients. Furthermore, we show that 31% of the disease-causing genes in the mitochondrial patient group were not included in the MitoCarta database, and therefore would have been missed with MitoCarta based gene-panels. We conclude that WES is preferable to panel-based approaches for both groups of patients, as the mitochondrial gene-list is not complete and mitochondrial symptoms can be secondary. Also, clinically and genetically heterogeneous disorders would require sequential use of multiple different gene panels. We conclude that WES is a comprehensive and unbiased approach to establish a genetic diagnosis in these patients, able to resolve multi-genic disease-causes.
线粒体疾病以临床症状和/或氧化磷酸化缺陷为特征,由线粒体基因中的致病变异引起。然而,这些基因中的一些致病变异可导致与其他神经肌肉疾病重叠的临床表现,而其他神经肌肉疾病也可由非线粒体基因中的致病变异引起。线粒体致病变异可在线粒体DNA(mtDNA)或1500个具有线粒体功能的核基因中的任何一个中发现。我们对117名患者(大多数为儿童)进行了两步下一代测序方法,这些患者的线粒体疾病病因可能或可能解释其表型。根据既定的临床和生化标准,共有86名患者患有线粒体疾病。其他31名患者有神经肌肉症状,其中少数存在线粒体遗传病因,但非线粒体遗传病因更有可能。对所有患者进行了mtDNA致病变异筛查,如被排除,则通过全外显子组测序(WES)进行分析。对变异进行过滤,以确定其致病性,并与有多个患病兄弟姐妹和/或近亲父母的家庭中的常染色体或X连锁隐性遗传模式兼容。对仅有一名患者的非近亲家庭,还在预定义的基因集中筛查常染色体和X连锁显性突变。我们在20%的患者队列中鉴定出mtDNA中的致病致病变异,在49%的患者队列中鉴定出核基因中的致病致病变异,总体检出率为68%。我们在两组中均鉴定出线粒体和非线粒体基因中的致病变异,显然,线粒体疾病患者中受影响的线粒体基因数量更多。此外,我们表明,线粒体患者组中31%的致病基因未包含在MitoCarta数据库中,因此基于MitoCarta的基因面板会遗漏这些基因。我们得出结论,对于两组患者,WES比基于面板的方法更可取。因为线粒体基因列表不完整,线粒体症状可能是继发性的。此外,临床和遗传异质性疾病需要依次使用多个不同的基因面板。我们得出结论,WES是一种全面且无偏倚的方法,可用于在这些患者中进行基因诊断,能够解析多基因疾病病因。