Infection, Immunity, Inflammation Programme, UCL GOS Institute of Child Health, London, UK.
Division of Neuropathology, UCL Institute of Neurology, London, UK.
Neuropathol Appl Neurobiol. 2019 Aug;45(5):495-512. doi: 10.1111/nan.12528. Epub 2019 Mar 11.
Juvenile idiopathic inflammatory myopathies have been recently reclassified into clinico-serological subgroups. Myopathological correlates of the subgroups are incompletely understood.
We studied muscle biopsies from 101 children with clinically and serologically defined juvenile idiopathic inflammatory myopathies from the UK JDM Cohort and Biomarker Study by applying the international JDM score tool, myopathological review and C5b-9 complement analysis.
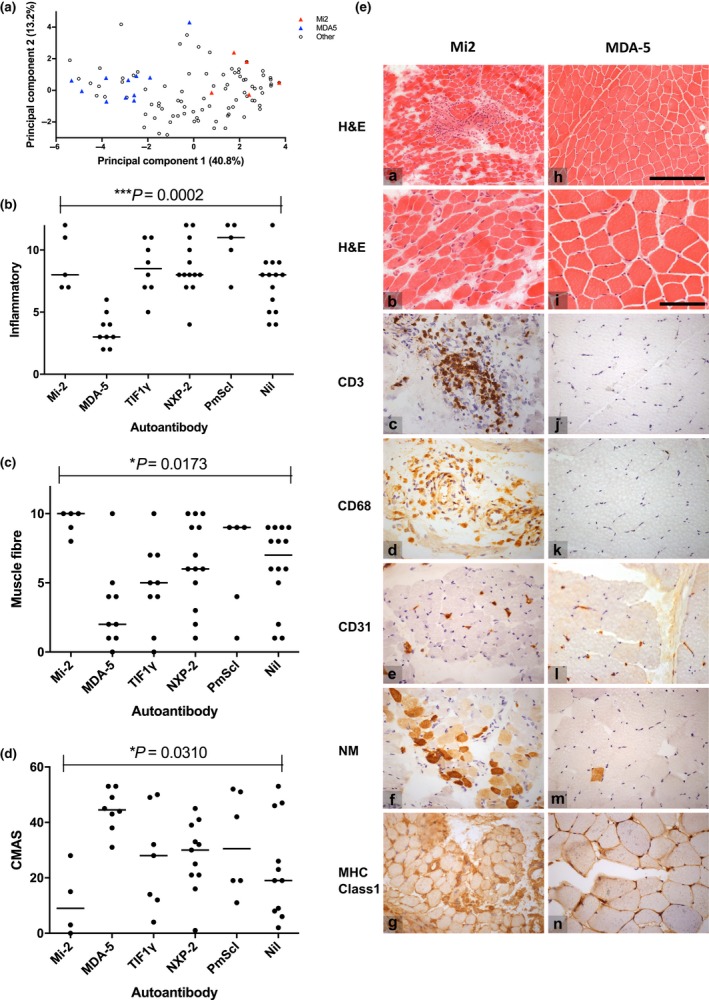
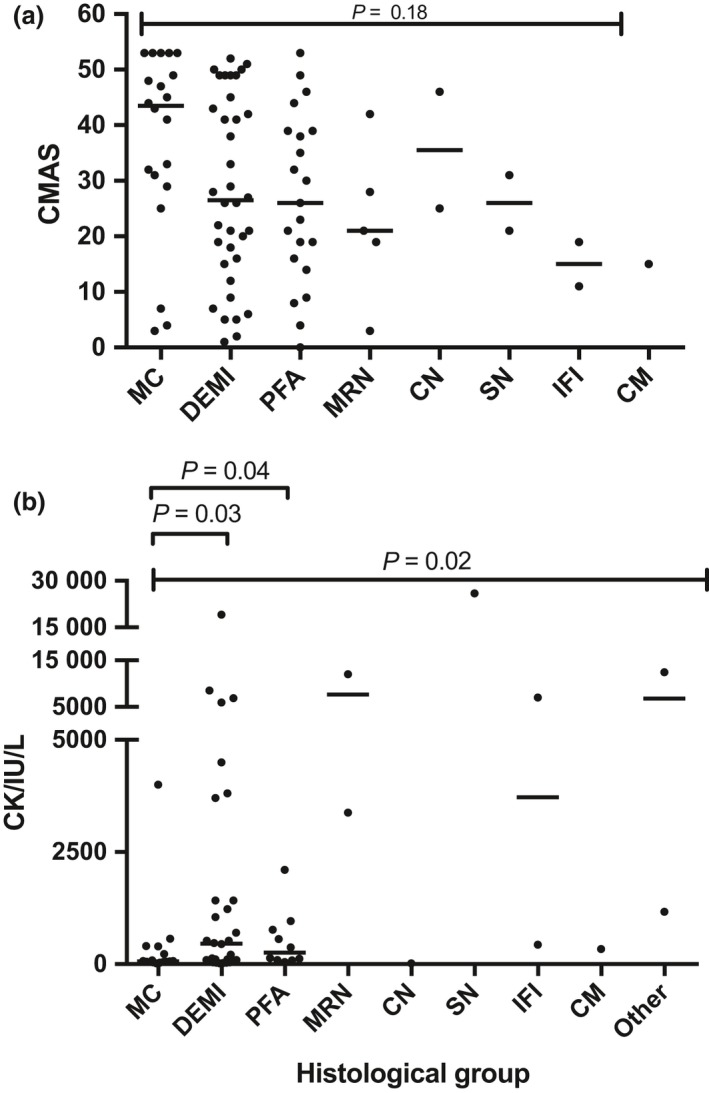
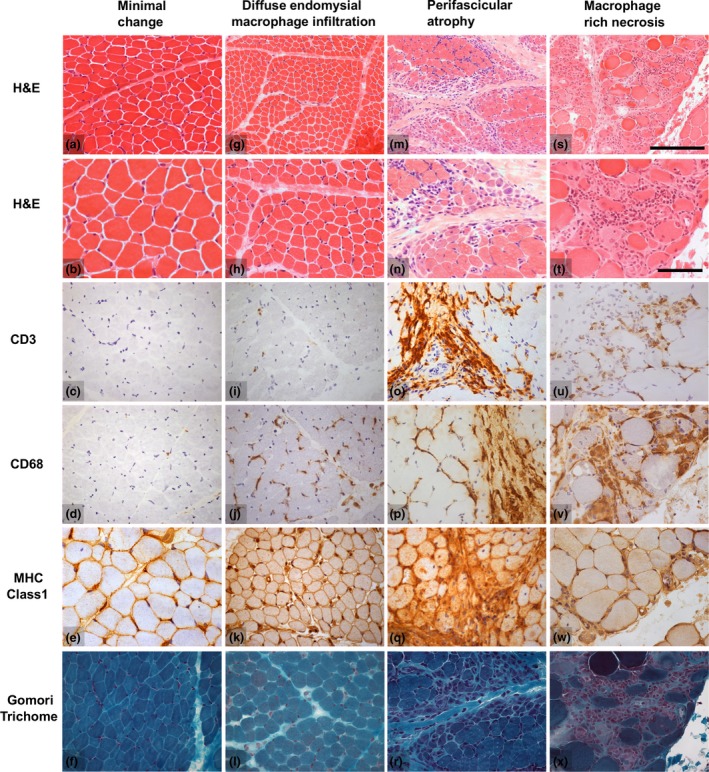
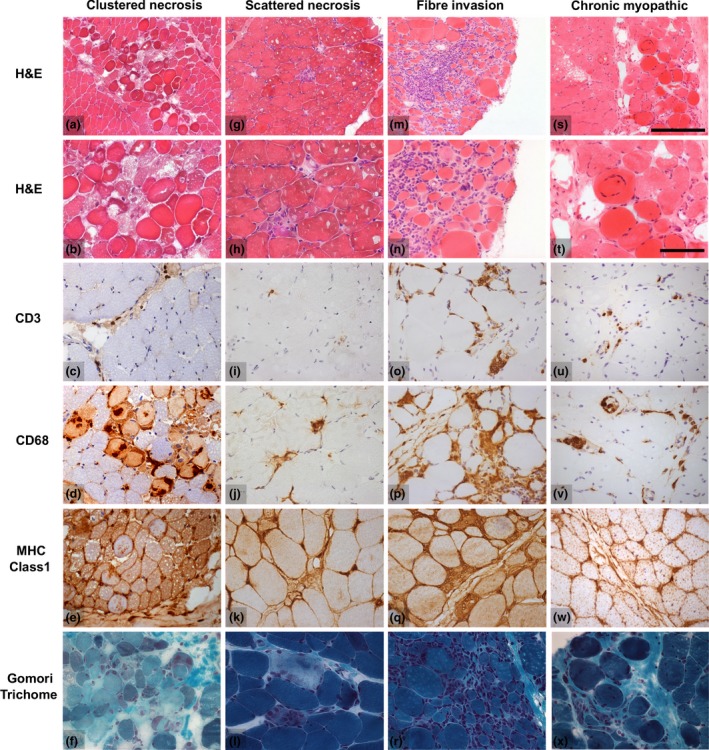
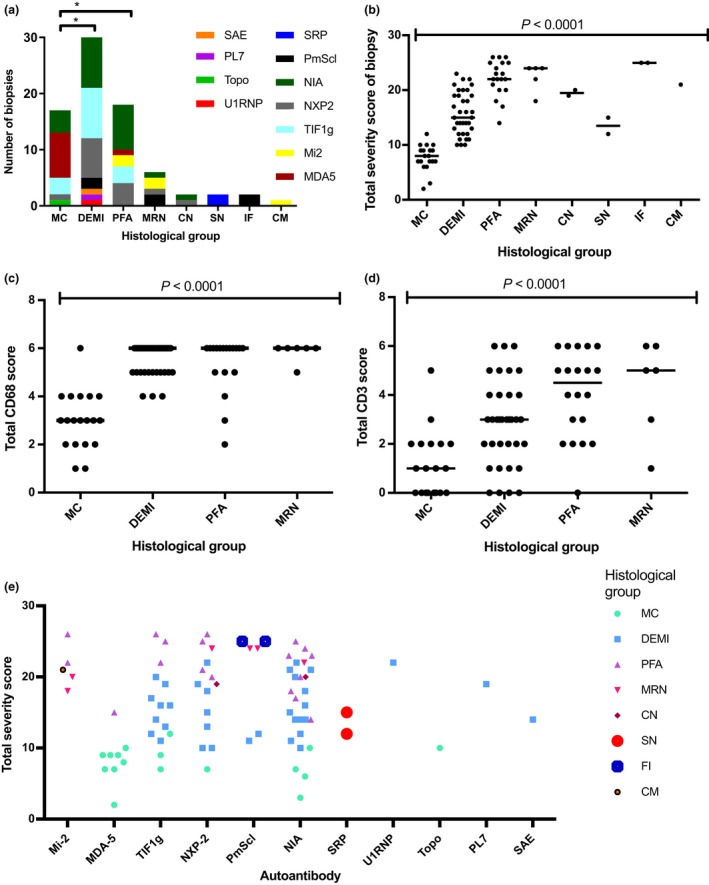
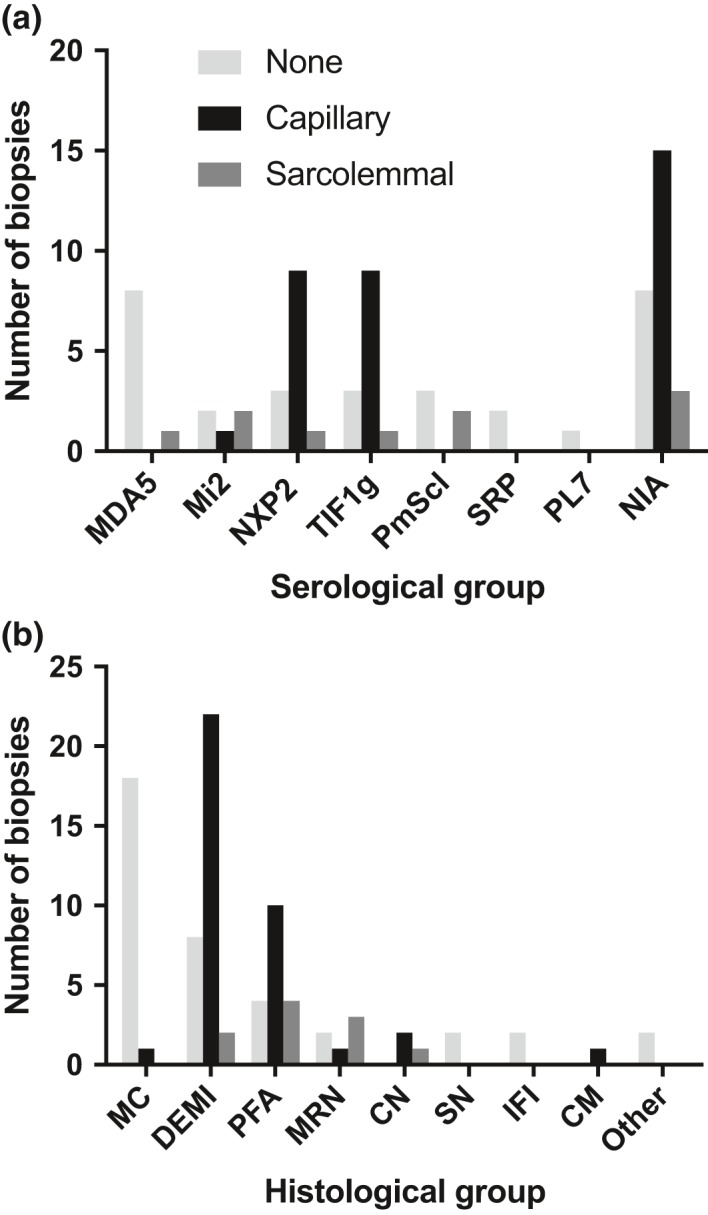
Autoantibody data were available for 90/101 cases with 18/90 cases positive for anti-TIF1γ, 15/90 anti-NXP2, 11/90 anti-MDA5, 5/90 anti-Mi2 and 6/90 anti-PmScl. JDM biopsy severity scores were consistently low in the anti-MDA5 group, high in the anti-Mi2 group, and widely distributed in the other groups. Biopsies were classified histologically as perifascicular atrophy (22/101), macrophage-rich necrosis (6/101), scattered necrosis (2/101), clustered necrosis (2/101), inflammatory fibre invasion (2/101), chronic myopathic change (1/101), diffuse endomysial macrophage infiltrates (40/101) and minimal change (24/101). MDA5 cases segregated with the minimal change group and showed no capillary C5b-9-deposition. The Mi2 group displayed high severity scores and a tendency towards sarcolemmal complement deposition. NXP2 and TIF1γ groups showed a variety of pathologies with a high proportion of diffuse endomysial macrophage infiltrates and a high proportion of capillary C5b-9 deposition.
We have shown that juvenile idiopathic inflammatory myopathies have a spectrum of histopathological phenotypes and show distinct complement attack complex deposition patterns. Both correlate in some cases with the serological subtypes. Most cases do not show typical histological features associated with dermatomyositis (e.g. perifascicular atrophy). In contrast, more than half show relatively mild histopathological changes.
最近,青少年特发性炎性肌病已重新分类为临床血清学亚组。这些亚组的肌肉病理相关性尚不完全清楚。
我们通过应用国际 JDM 评分工具、肌肉病理检查和 C5b-9 补体分析,对来自英国 JDM 队列和生物标志物研究的 101 例经临床和血清学定义的青少年特发性炎性肌病患儿的肌肉活检进行了研究。
90/101 例病例可获得自身抗体数据,其中 18/90 例抗 TIF1γ 阳性、15/90 例抗 NXP2 阳性、11/90 例抗 MDA5 阳性、5/90 例抗 Mi2 阳性和 6/90 例抗 PM-Scl 阳性。抗 MDA5 组的 JDM 活检严重程度评分始终较低,抗 Mi2 组较高,而其他组则广泛分布。活检根据组织学表现分为束周萎缩(22/101)、巨噬细胞丰富性坏死(6/101)、散在性坏死(2/101)、簇状坏死(2/101)、炎性纤维浸润(2/101)、慢性肌病性改变(1/101)、弥漫性内肌层巨噬细胞浸润(40/101)和微小改变(24/101)。MDA5 病例与微小改变组分离,无毛细血管 C5b-9 沉积。Mi2 组显示出较高的严重程度评分和肌膜补体沉积的趋势。NXP2 和 TIF1γ 组表现出多种病理改变,弥漫性内肌层巨噬细胞浸润比例高,毛细血管 C5b-9 沉积比例高。
我们已经表明,青少年特发性炎性肌病具有一系列组织病理学表型,并显示出不同的补体攻击复合物沉积模式。在某些情况下,两者与血清学亚型相关。大多数病例没有显示出与皮肌炎相关的典型组织学特征(例如束周萎缩)。相反,超过一半的病例显示相对轻微的组织病理学变化。