Department of Animal and Plant Sciences, University of Sheffield, Sheffield UK, UK.
INSIGNEO Institute for In Silico Medicine, University of Sheffield, Sheffield UK, UK.
Bioinformatics. 2019 Jul 1;35(13):2243-2250. doi: 10.1093/bioinformatics/bty946.
In clinical trials, individuals are matched using demographic criteria, paired and then randomly assigned to treatment and control groups to determine a drug's efficacy. A chief cause for the irreproducibility of results across pilot to Phase-III trials is population stratification bias caused by the uneven distribution of ancestries in the treatment and control groups.
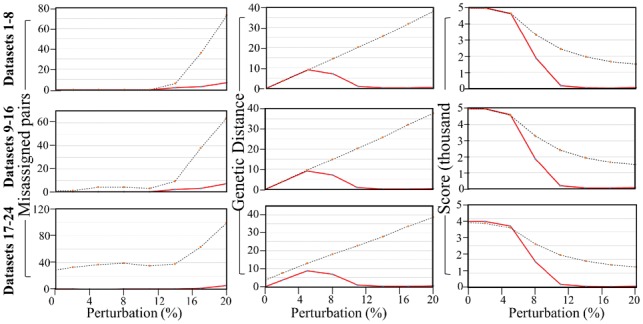
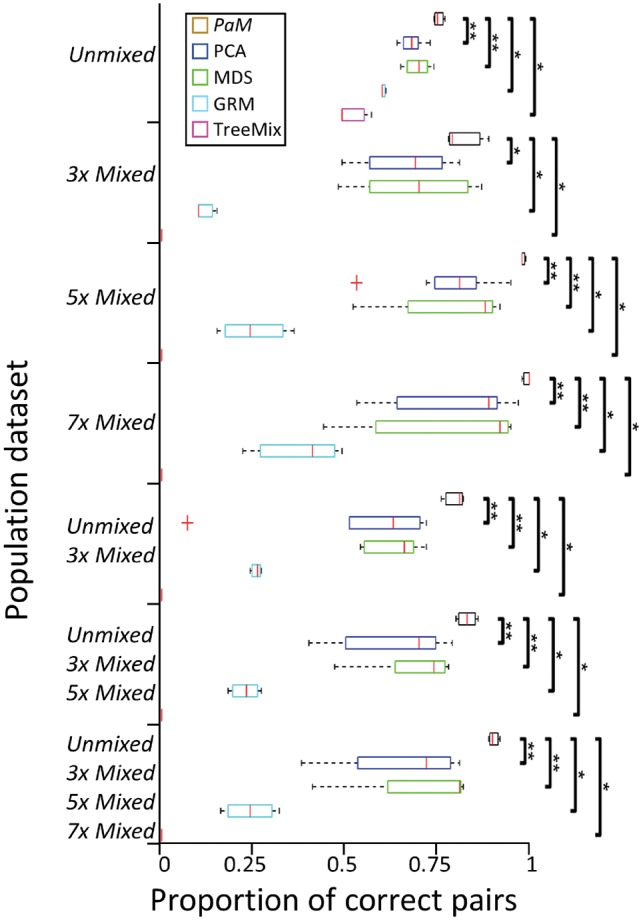
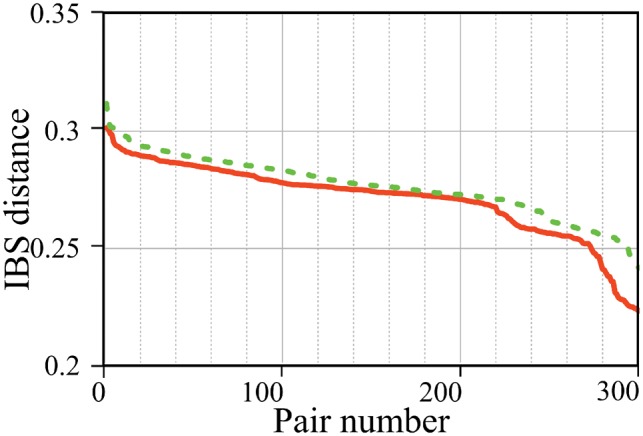
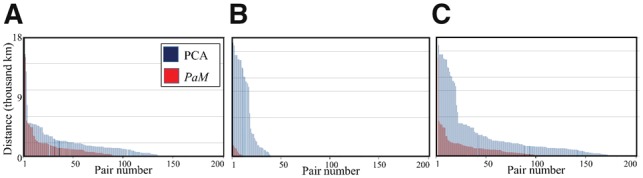
Pair Matcher (PaM) addresses stratification bias by optimizing pairing assignments a priori and/or a posteriori to the trial using both genetic and demographic criteria. Using simulated and real datasets, we show that PaM identifies ideal and near-ideal pairs that are more genetically homogeneous than those identified based on competing methods, including the commonly used principal component analysis (PCA). Homogenizing the treatment (or case) and control groups can be expected to improve the accuracy and reproducibility of the trial or genetic study. PaM's ancestral inferences also allow characterizing responders and developing a precision medicine approach to treatment.
PaM is freely available via Rhttps://github.com/eelhaik/PAM and a web-interface at http://elhaik-matcher.sheffield.ac.uk/ElhaikLab/.
Supplementary data are available at Bioinformatics online.
在临床试验中,个体通过人口统计学标准进行匹配,配对后再随机分配到治疗组和对照组,以确定药物的疗效。导致从试点到 III 期试验的结果不可重现的一个主要原因是,由于治疗组和对照组中祖源的分布不均,导致了群体分层偏差。
配对匹配器(PaM)通过使用遗传和人口统计学标准,在试验之前和/或之后优化配对分配,解决了分层偏差问题。使用模拟和真实数据集,我们表明 PaM 可以识别出理想和近乎理想的配对,这些配对比基于竞争方法(包括常用的主成分分析(PCA))识别出的配对在遗传上更同质。使治疗(或病例)组和对照组同质化有望提高试验或遗传研究的准确性和可重复性。PaM 的祖先推断还允许对响应者进行特征描述,并开发一种针对治疗的精准医学方法。
PaM 可通过 R 免费获得https://github.com/eelhaik/PAM 以及一个网络界面http://elhaik-matcher.sheffield.ac.uk/ElhaikLab/。
补充数据可在生物信息学在线获得。