Institute for Human Genetics, Institute for Health and Computational Sciences, Department of Biostatistics and Epidemiology, Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco, California 94143, USA.
Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02142, USA.
Genome Res. 2018 Dec;28(12):1812-1825. doi: 10.1101/gr.240390.118. Epub 2018 Nov 16.
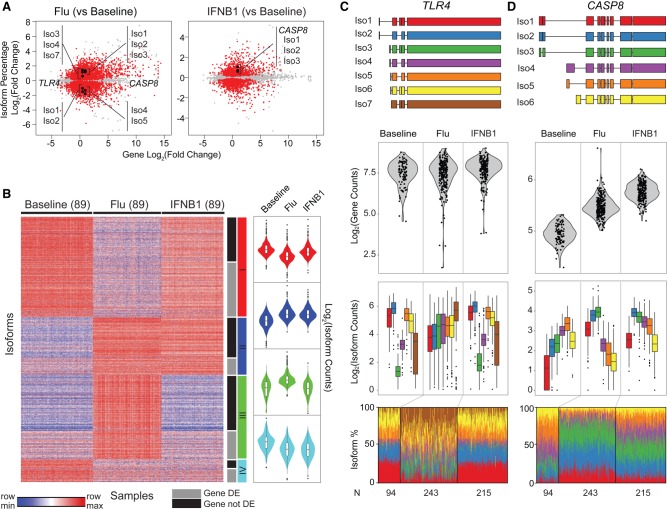
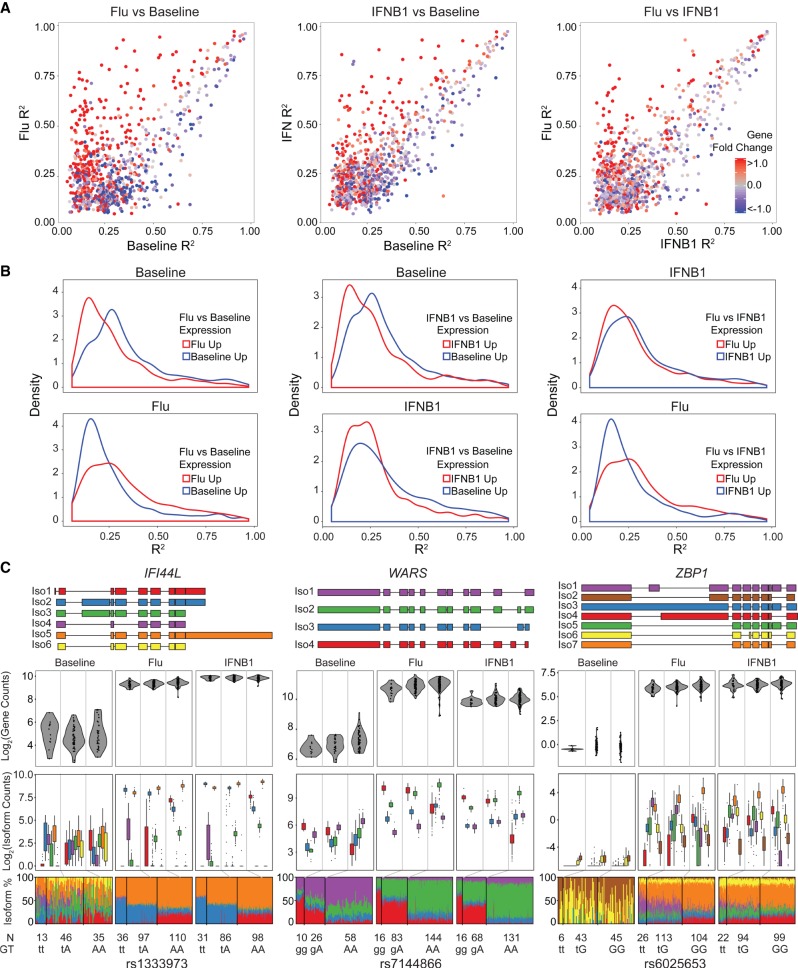
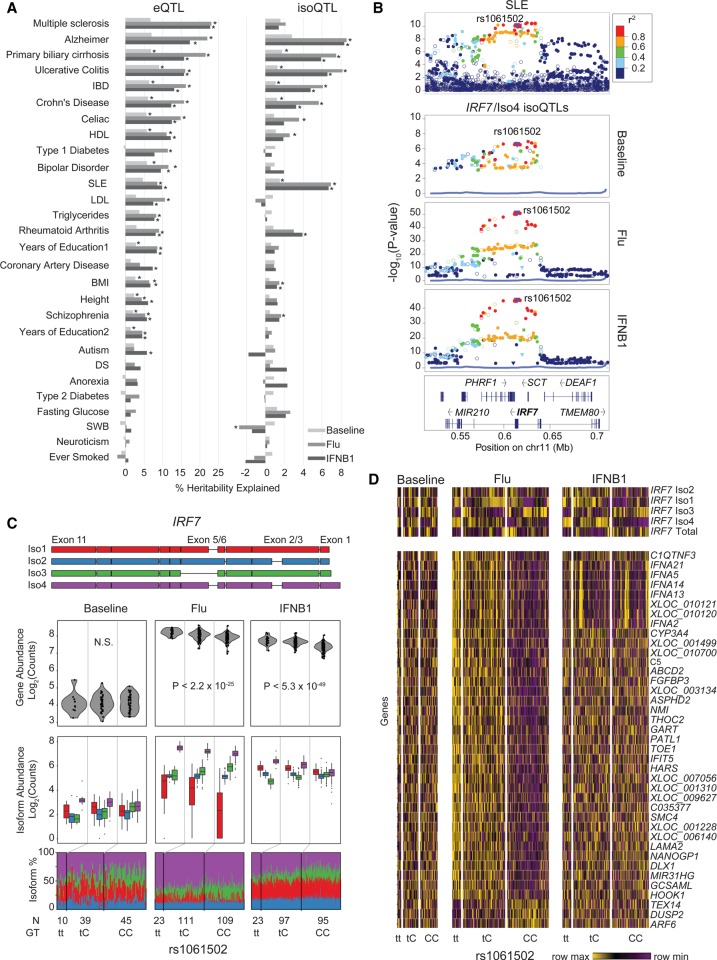
While genetic variants are known to be associated with overall gene abundance in stimulated immune cells, less is known about their effects on alternative isoform usage. By analyzing RNA-seq profiles of monocyte-derived dendritic cells from 243 individuals, we uncovered thousands of unannotated isoforms synthesized in response to influenza infection and type 1 interferon stimulation. We identified more than a thousand quantitative trait loci (QTLs) associated with alternate isoform usage (isoQTLs), many of which are independent of expression QTLs (eQTLs) for the same gene. Compared with eQTLs, isoQTLs are enriched for splice sites and untranslated regions, but depleted of sequences upstream of annotated transcription start sites. Both eQTLs and isoQTLs explain a significant proportion of the disease heritability attributed to common genetic variants. At the locus, we shed light on the function of the gene and how two frequent, highly differentiated haplotypes with intermediate frequencies could be maintained by balancing selection. At baseline and following type 1 interferon stimulation, the major haplotype is associated with low expression caused by nonsense-mediated decay, while the minor haplotype, known to increase Crohn's disease risk, is associated with high expression. In response to influenza infection, we found two uncharacterized isoforms expressed from the major haplotype, likely the result of multiple perfectly linked variants affecting the transcription and splicing at the locus. Thus, genetic variants at a single locus could modulate independent gene regulatory processes in innate immune responses and, in the case of , may confer a historical fitness advantage in response to virus.
虽然已知遗传变异与刺激免疫细胞中的整体基因丰度有关,但它们对替代异构体使用的影响知之甚少。通过分析 243 个人单核细胞衍生树突状细胞的 RNA-seq 图谱,我们发现了数千种在流感感染和 I 型干扰素刺激下合成的未注释异构体。我们确定了超过一千个与替代异构体使用(isoQTL)相关的数量性状基因座(QTL),其中许多与同一基因的表达 QTL(eQTL)无关。与 eQTL 相比,isoQTL 在剪接位点和非翻译区富集,但在注释转录起始位点上游的序列中则减少。eQTL 和 isoQTL 都解释了归因于常见遗传变异的疾病遗传率的很大一部分。在 基因座上,我们阐明了该基因的功能以及两种常见、高度分化的中等频率单倍型如何通过平衡选择得以维持。在基线和 I 型干扰素刺激后,主要单倍型与由于无意义介导的衰变导致的低 表达相关,而已知增加克罗恩病风险的次要单倍型与高 表达相关。在流感感染后,我们从主要单倍型中发现了两种未表征的异构体,可能是影响该基因座转录和剪接的多个完美连锁变异的结果。因此,单个基因座的遗传变异可以调节固有免疫反应中的独立基因调控过程,而就 而言,在应对病毒时可能赋予历史适应优势。