Department of Biochemistry, Faculty of Biological Sciences, Quaid-i-Azam University, University Road, Islamabad, Post code 45320, Pakistan.
Institute of Biomedical and Genetic Engineering (IBGE) Islamabad, Islamabad, 44000, Pakistan.
J Biomed Sci. 2018 Nov 17;25(1):82. doi: 10.1186/s12929-018-0481-x.
Osteogenesis imperfecta (OI) is a clinically and genetically heterogeneous disease with skeletal fragility and variable extra-skeletal manifestations. To date several point mutations in 18 different genes causing different types of OI have been identified. Mutations in WNT1 compromise activity of the osteoblasts leading to disturbed bone mass accrual, fragility fractures and progressive skeletal abnormalities. The present study was conducted to determine the underlying genetic cause of an autosomal recessive skeletal dysplasia in a large consanguineous family from Chinute, Pakistan.
Blood was collected from 24 individuals of affected family along with clinical data. Homozygosity mapping was performed to confirm consanguinity. SNPs were identified, followed by whole exome and Sanger sequencing. In silico characterization of WNT1 mutation was performed using multiple platforms.
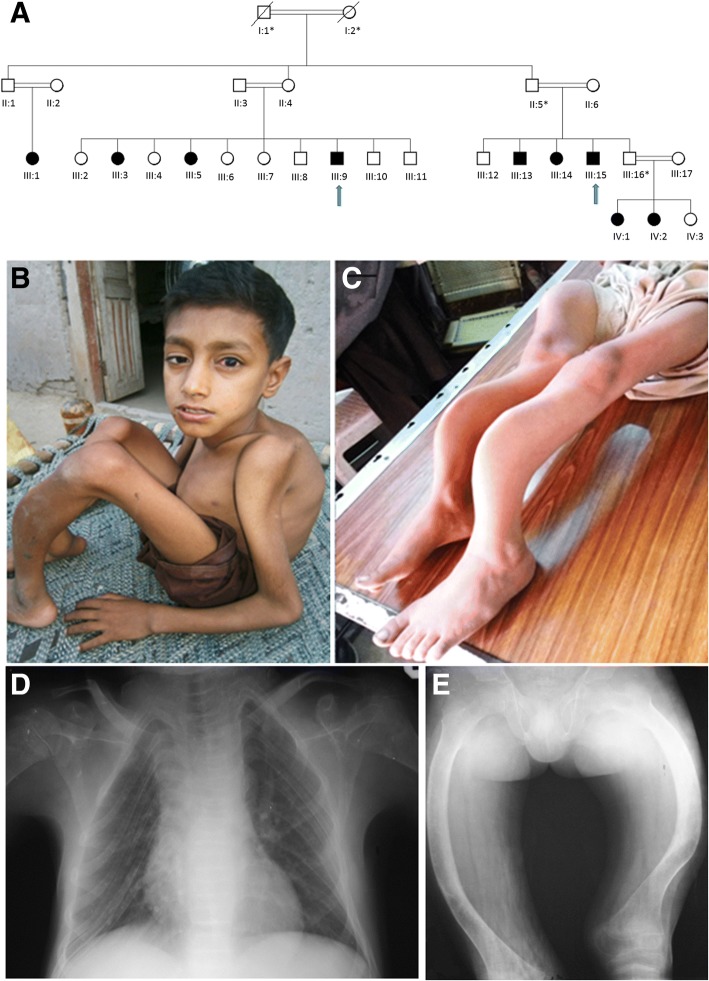
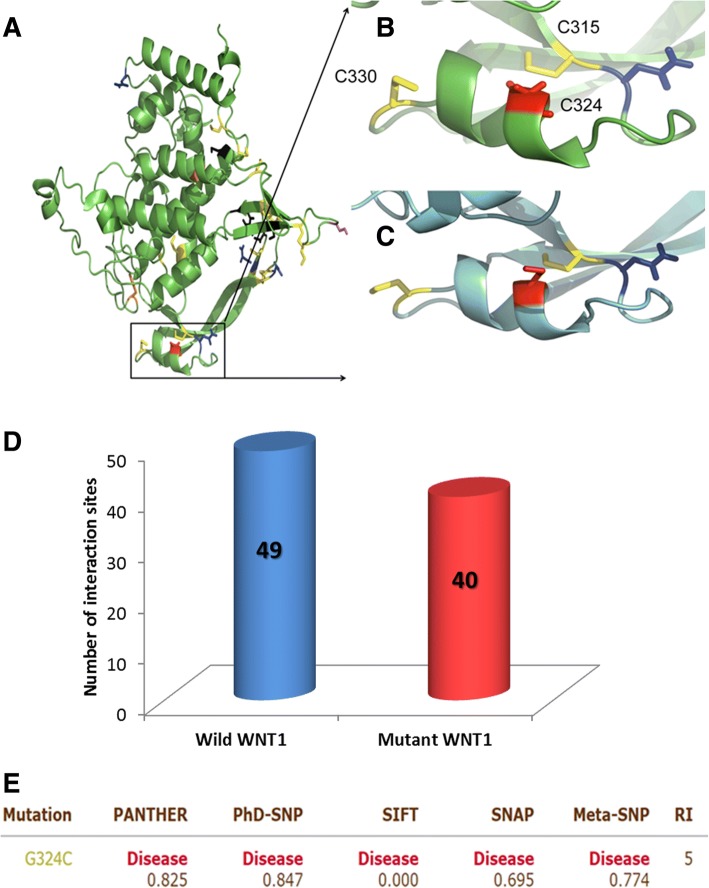
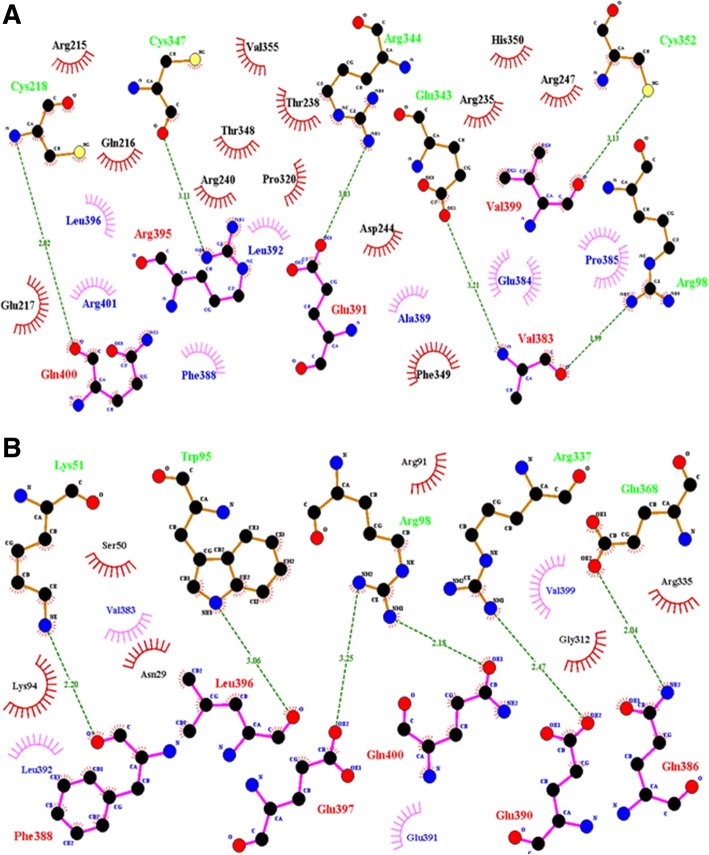
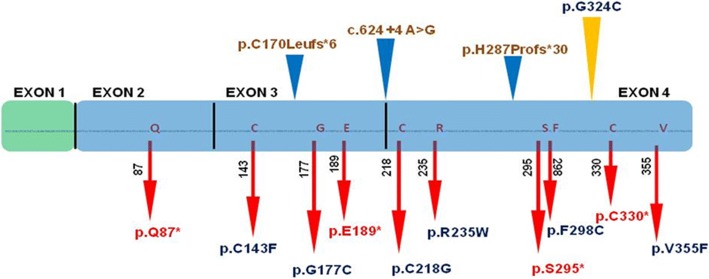
Nine affected family members exhibited severe bone deformities, recurrent fractures, short stature and low bone mineral density. SNP array data revealed homozygous segments > 1 Mb in length accounting for 2.1-12.7% of the genome in affected individuals and their siblings and a single 6,344,821 bp homozygous region in all affected individuals on chromosome 12q12-q13. This region includes two potential OI candidate genes WNT1 and VDR. We did whole-exome sequencing for both genes in two patients and identified a novel damaging missense mutation in exon 4 of WNT1: c.1168G > T (NM_005430) resulting in p.G324C. Sanger sequencing confirmed segregation of mutation with the disease in family.
We report a novel mutation responsible for OI and our investigation expands the spectrum of disease-causing WNT1 mutations and the resulting OI phenotypes.
成骨不全症(OI)是一种临床和遗传异质性疾病,具有骨骼脆弱和可变的骨骼外表现。迄今为止,已经在 18 个不同的基因中发现了导致不同类型 OI 的几个点突变。WNT1 突变会削弱成骨细胞的活性,导致骨量积累异常、脆性骨折和进行性骨骼异常。本研究旨在确定来自巴基斯坦 Chinute 的一个大型近亲家族的常染色体隐性骨骼发育不良的潜在遗传原因。
采集了 24 名受影响家族成员的血液和临床数据。进行了同系性作图以确认近亲关系。确定了 SNP 后,进行了全外显子和 Sanger 测序。使用多个平台对 WNT1 突变进行了计算机模拟分析。
9 名受影响的家庭成员表现出严重的骨骼畸形、反复骨折、身材矮小和低骨密度。SNP 数组数据显示,受影响个体及其兄弟姐妹的基因组中存在长度超过 1 Mb 的纯合片段,占 2.1-12.7%,所有受影响个体在染色体 12q12-q13 上都存在一个 6344821 bp 的纯合区域。该区域包括两个潜在的 OI 候选基因 WNT1 和 VDR。我们对两名患者的这两个基因进行了全外显子组测序,发现了 WNT1 外显子 4 中的一个新的有害错义突变:c.1168G>T(NM_005430)导致 p.G324C。Sanger 测序证实该突变在家族中与疾病共分离。
我们报告了一个导致 OI 的新突变,我们的研究扩大了 WNT1 突变导致的疾病谱和由此产生的 OI 表型。